










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El Sarcoma de Ewing (SE) forma parte de los tumores infrecuentes de cabeza y cuello, Fuedescrito por primera vez en 1918 por James Ewing, pero no fue hasta 1921 que se describió su origen histogenetico,1son tumores muy agresivos provocando metástasis precozmente, por lo que obliga a dar un pronóstico muy desfavorable; forman parte de una gran familia de tumores cuyo origen se encuentra en las células embrionarias, el SE comparte características con los tumores neuroectodérmicos primitivos.1,2
Normalmente se desarrolla en los tejidos musculares y óseos, pero puede desarrollarse en cualquier localización. 3 Se encuentra dentro de los tumores de células pequeñas, redondas y azules, junto con otros tumores, como los linfomas, tumores neuroectodérmicos primitivos, rabdomiosarcoma, neuroblastoma, entre otros en los cuales debemos pensar como diagnóstico diferencial. (1,3
Este tipo de tumor aparece con mayor frecuencia en niños y adultos jóvenes, muchos autores señalan entre los 8 y los 20 años, pero pueden presentarse a cualquier edad con predominio en el sexo masculino en una proporción de 1,5:11. (4El pronóstico depende de la existencia de metástasis debido a que el sarcoma de Ewing es sumamente maligno y causa metástasis al hueso y pulmón por lo que la supervivencia es relativamente baja.
Con este artículo pretendemos hacer una descripción del caso de una niña con Sarcoma de Ewingnasosinusal, siendo este tipo de tumor una enfermedad rara en Gabón y a nivel mundial, nos dimos a la tarea de realizar una revisión de la literatura para conocer incidencia, etiología, pronóstico, y tratamiento multidisciplinario.
Caso clínico
Presentamos una niña de 4 años de edad con antecedentes personales de buena salud, de procedencia rural; es traída a consulta en mayo de 2017 presentando una masa tumoral en la fosa nasal derecha que provocaba ligera obstrucción nasal, nunca tuvo episodios de epistaxis o rinorrea, ni antecedentes de trauma en la región facial o algún tipo de infección viral o catarral.
Al practicar el examen físico encontramos una masa sólida, de forma elíptica entre el cuerpo del hueso malar y la parte anterosuperior del hueso maxilar derecho. Existía límite entre la lesión y el músculo circundante y no infiltraba la piel pero formaba un ligero relieve subcutáneo hacia el interior de fosa nasal del mismo lado. En la exploración al tacto en la región intraoral encontramos una pequeña masa firme, móvil e indolora en la encía del fondo del vestíbulo de la arcada dentaria superior derecha. No encontramos ganglios en cuello ni otro signo alarmante al examen físico general.(Fig. 1)
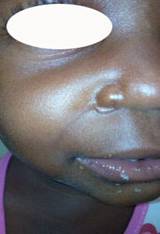
Una vez realizado el examen físico y exámenes de hematología indicamos una Resonancia Magnética (RM),en su informenos muestra la presencia de una masa tumoral hacia el piso nasal derecho, de gran intensidad, isointenso, con integridad de las estructuras vecinasóseas.
Realizamos cirugía incisional para tomar biopsia, haciendo un abordaje intraoral (Fig. 2). Encontramos una masa tumoral, redondeada, con elementos solidos de consistencia dura, y friable hacia la zona de implantación, constituida, en su mayor parte, por una proliferación compacta de células pequeñas. (Fig. 3)
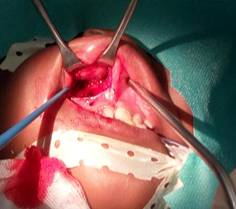
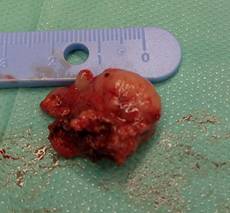
La biopsia no es concluyente, informa linfoma maligno no Hopkins o sarcoma de Ewing a confrontar con la clínica, indicamos marcadores tumorales para asegurar diagnóstico, se le realizaron CD 45 Dako clon 2b11 mas Pd7/26 para descartar tumores de naturaleza linfoidea el cual fue negativo, se realizó Dako CD99 y anticuerpos anti synaptophisine (Dako clon 19) buscando la presencia de tumores epiteliales y neuroblastomas lo cual dio positivo, se interpretó como un Sarcoma de Ewing de fosa nasal.
Referida al centro oncológico comenzó protocolo de tratamiento con:
Premedicación: Solumedrol 20 mg y Ondasetron 4 mg intravenoso 30 minutos antes de Oncovin.
Oncovin 0.8 mg diluido en 50 ml de solución salina a pasar intravenoso en 15 minutos.
Glucosa 5% 500 ml con cyclosphosphamida 660 mg diluido en el suero de glucosa a pasar en 1 hora.
Adriblastina 40 mg intravenoso diluido lento.
Transcurrido dos meses del tratamiento encontramos cambios positivos favorables de buena evolución, durante el examen físico no encontramos signos de agresión o lisis ósea, indicamos reconsulta cada mes para control clínico. Un año después decidimos realizar cirugía reconstructiva para reparar la función fisiológica respiratoria de la fosa nasal.
Discusión
El sarcoma de Ewing es la segunda neoplasia ósea maligna más frecuente en pediatría; sin embargo, es infrecuente en tejidos blandos con una baja tasa de incidencia entre el 1%-3% incluyendo el SEnasosinusal, sin dejar de mencionar a los tumores neuroectodérmicos primarios que son tumores de células pequeñas y redondas derivadas del tejido blando, y que pertenecen a la familia del sarcoma de Ewing. (5,6
El diagnóstico está basado en los elementos clínicos, radiológicos y patológicos. Los síntomas y signos son variables pueden aparecer epistaxis frecuentes sobre todo en fases avanzadas de la enfermedad, obstrucción nasal, rinorrea purulenta, si existe lisis o destrucción ósea el paciente puede referir dolor a nivel facial, nuestra paciente solo presentaba obstrucción nasal unilateral derecha.
Histológicamente el sarcoma de Ewing tiene origen tanto mesodérmico como ectodérmico por lo tanto su clasificación es difícil. Se encuentra entre los tumores de células redondas pequeñas de la infancia, caracterizado por la presencia de células redondas de pequeño tamaño con núcleo hipercromático, bordes bien definidos y ausencia de material intercelular; existe abundante glucógeno citoplasmático. (7
La etiología exacta sigue siendo desconocida, algunos autores plantean que este tumor presenta una anomalía citogenética que lo diferencia de otros tumores pediátricos como el neuroblastoma y el rabdomiosarcoma, no relacionado con síndromes congénitos o síndromes neoplásicos; la literatura revisada menciona que esta familia de sarcomas comparte una única secuencia de especifica de translocación que implica a los cromosomas 11 y 22, t(11;22)(q24;q12), la cual resulta en la expresión de una proteína quimérica, EWSR1-FLI1. Esta translocación está presente en el 85-95% de los casos que han presentado tumores de la familia del Sarcoma de Ewing. (6,7
Los marcadores tumorales son esenciales para poder hacer diagnóstico diferencial con otros tumores que comparten características similares al SE; se hace necesario evaluar la inmunohistoquimica de la Vimentina cuando es positiva habla a favor del melanoma mucoso y el rabdomiosarcoma, para descartar el carcinoma neuroendocrino, rabdomiosarcoma y adenoma pituitario se indican marcadores CD56,positivo en estos casos; la proteína S100 es positiva hasta un 30 % del SE, además del CD99 que es considerado el marcador mássensible, debido a una fuerte y difusa reactividad membranosa CD99 del Sarcoma de Ewing. 8
El diagnóstico diferencial de los tumores en la cavidad nasal y los senos paranasales con afección o no intracraneal incluye los meningiomas o metástasis, extensión directa de tumores de la base del cráneo o de la nasofaringe descartando el estesioneuroblastoma, carcinoma nasofaríngeo, melanoma, rabdomiosarcoma o linfoma, en ocasiones se incluye el angiofibroma nasal. 4
Las características más comunes a nivel de tomografía computarizada es la presencia de una masacon densidad de partes blandas que capta medio de contraste en forma irregular; y a nivel de RM, lesión isointensa o levemente hiperintesa en T1 e hiperintensa en T26. Hallazgos que encontramos en la RM practicada a nuestra paciente. (4,8
En la bibliografía consultada, el tratamiento para la familia de los tumores de Ewing está basado en la administración de una dosis alta de quimioterapia para el control sistémico de la enfermedad, seguido de un control local, que incluye amplia resección o irradiación del tejido afecto. Debido a la agresividad de estos tumores se propone un tratamiento combinado, consistente en una resección quirúrgica con quimio y radioterapia adyuvantes. Se puede utilizar también quimioterapia neo adyuvante para facilitar la resección quirúrgica y disminuir los riesgos intraoperatorios. La quimioterapia es muy relevante, ya que produce una importante mejoría en el pronóstico de estos pacientes, garantizando la supervivencia. (4,6,8,9
La EuropeanInter group Cooperative recomienda 14 ciclos de etoposido, vincristina, ifosfamida y adriamicina. De acuerdo con este protocolo la quimioterapia se repite cada 3 semanas. Desde la introducción de la quimioterapia la supervivencia ha mejorado significativamente, con una sobrevida de 5 años. Los sitios más frecuentes de metástasis corresponden a pulmón, hígado y medula ósea. (9,10
Conclusiones
El sarcoma de Ewing es un tumor poco frecuente, se presenta habitualmente en niños y jóvenes con un comportamiento altamente agresivo, son de rápida y extensa diseminación. La presencia de células pequeñas redondas y azules más el estudio inmunohistoquimico nos confirma el diagnostico. Para el tratamiento aún no existe un consenso y protocolos universalmente aceptados, sin embargo, se requiere un enfoque multidisciplinario, con un enfrentamiento agresivo y combinado con cirugía y radio-quimioterapia. El buen estado general del paciente en el momento del diagnóstico resultó muy favorable en su respuesta al tratamiento oncológico y evolución, a un año de efectuado el diagnóstico y manejo inicial, no se encuentra signos clínicos ni imagenológicos de persistencia o recidiva tumoral.

