










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión impresa ISSN 0034-7515
Rev Cubana Farm vol.48 no.2 Ciudad de la Habana abr.-jun. 2014
ARTÍCULO ORIGINAL
Estabilidad de supositorios de naproxeno para uso infantil y adulto
Stability of Naproxen suppositories for children and adults
DrC. Yania Suárez Pérez,I Oscar García Pulpeiro,II Mabert Placert ÁlvarezIII
I Instituto de Farmacia y Alimentos. Universidad de la Habana. La Habana, Cuba.
II Empresa Roberto Escudero Díaz, QUIMEFA. La Habana, Cuba.
III Centro de Inmunología Molecular. La Habana, Cuba.
RESUMEN
Introducción : el naproxeno en supositorios para uso infantil y adulto constituye una de las líneas de investigación en desarrollo de la Empresa "Roberto Escudero Díaz". Los estudios de estabilidad son una parte indispensable para el registro de una nueva formulación.
Objetivo: determinar la estabilidad de los supositorios de naproxeno para uso infantil y adulto, teniendo en cuenta la estabilidad física y química del analito en las nuevas formulaciones.
Métodos: se realizó el estudio de estabilidad para formulaciones en fase de desarrollo de supositorios de naproxeno para uso infantil y adulto, teniendo en cuenta la metodología propuesta por el Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Se emplearon para cada dosis evaluada, supositorios de tres lotes pilotos envasados en tiras de aluminio termosellables. Se almacenaron los supositorios a temperatura de refrigeración (2-8 °C) y ambiente (30 ± 2 °C) durante un año, y se les realizaron muestreos a los 0, 1, 3, 6 y 12 meses de elaborados. Para el análisis de la estabilidad química, se consideraron los resultados del contenido de naproxeno obtenidos por volumetría de neutralización y por cromatografía líquida de alta resolución, así como la determinación de los posibles productos de degradación por cromatografía en capa delgada.
Resultados: se demostró la adecuada estabilidad física de los supositorios de ambas dosis, independientemente de la temperatura de almacenamiento durante 12 meses, ya que se mantuvieron inalteradas las características organolépticas y el peso. Aunque el tiempo de liquefacción disminuyó durante el almacenamiento, siempre fue inferior al límite establecido. Con el método por cromatografía líquida de alta resolución, se detectaron pequeños cambios en la concentración de analito, por lo que este método fue superior para el seguimiento de la estabilidad química que la volumetría de neutralización. No se detectaron productos de degradación en los supositorios por ninguna de las técnicas cromatográficas utilizadas.
Conclusiones: los supositorios fueron estables desde el punto de vista físico y químico a temperatura de refrigeración (2-8 °C) y ambiente (30 ± 2 °C) durante 12 meses.
Palabras clave: naproxeno, supositorios, estabilidad.
ABSTRACT
Introduction: Naproxen suppository for children and adults is one of the developing research lines of "Roberto Escudero Diaz" drug production enterprise. The stability studies are indispensable for the registration of a new formulation.
Objective: to determine the stability of Naproxen suppositories for children and adults, taking the physical and chemical stability of the analyte into account in the new formulations.
Methods: pursuant to the methodology of the Center for the State Quality Control of Drugs (CECMED), the stability study for developing formulations was conducted in Naproxen suppositories for children and adults. These products packaged in heat-seal aluminum blister packs from three pilot batches were used for each evaluated dose. They were stored at 2 to 8 °C refrigeration and at air temperature of 30 ± 2 °C during one year, and sampled at 0, 1, 3, 6 and 12 months after preparation. For the analysis of the chemical stability, the Naproxen content determined by the neutralizing volumetry and high performance liquid chromatography tests and the possible degradation products identified in the thin layer chromatography test were taken into account.
Results: this study proved the adequate physical stability of suppositories at both doses regardless of the storage temperatures during 12 months because their organoleptic characteristics and weight remained unchanged. Although the liquefaction time decreased under the storage conditions, it was lower than the set limit. The high performance liquid chromatography detected slight changes in the analyte concentration, so this method was better for the chemical stability analysis than the neutralization volumetry method. The chromatographic techniques did not detect any degradation product in the suppositories.
Conclusions: the suppositories were physically and chemically stable at a refrigerating temperature of 2 to 8 °C and at air temperature of 30 ± 2 °C during 12 months.
Key words: Naproxen, suppositories, stability.
INTRODUCCIÓN
El naproxeno se comercializa en el mundo en diversas formas farmacéuticas, tales como: inyectables, cremas, cápsulas, tabletas y supositorios,1-5 teniendo en cuenta sus múltiples aplicaciones como potente antiinflamatorio en diversas enfermedades: otitis, dolores menstruales, artritis, tendinitis, gota, síndromes de la columna vertebral, estados postraumáticos, migraña, estados posoperatorios, ondontalgias y como coadyuvante es estados gripales.6 En Cuba se produce solamente en forma de tabletas.7
El desarrollo y registro del naproxeno en supositorios para uso infantil y adulto constituye una de las líneas de investigación priorizada en la empresa "Roberto Escudero Díaz", único centro que se dedica a la producción de este tipo de forma farmacéutica en el país. Esta investigación se fundamenta en que por administración rectal, los efectos adversos del naproxeno son menores que por vía oral. Al igual que otros analgésicos antiinflamatorios no esteroideos (AINEs), produce gran irritación en la mucosa gástrica.8
Como parte del conjunto de investigaciones que se requiere para el registro de un nuevo producto, se destacan por su importancia los estudios de estabilidad, empleando métodos adecuados a fin de comprobar la estabilidad física, química y microbiológica para establecer el período de validez y las condiciones de almacenamiento propuestos por el fabricante para la etapa de comercialización.9
Los requerimientos para las pruebas de estabilidad han sido armonizados por los organismos reguladores a nivel internacional10 y a nivel nacional,11-13 en lo referido a: tipo de estudio de estabilidad, condiciones de almacenamiento, tipo de lotes a analizar, frecuencia de muestreo y duración del ensayo.
Teniendo en cuenta estos antecedentes, se propone como objetivo determinar la estabilidad de los supositorios de naproxeno para uso infantil y adulto, teniendo en cuenta la estabilidad física y química del analito en las nuevas formulaciones.
MÉTODOS
Se emplearon supositorios de naproxeno elaborados a escala piloto en la Empresa "Roberto Escudero Díaz", para uso infantil (dosis: 50 mg; lotes: 10001, 10002 y 10003) y adulto (dosis: 500 mg; lotes: 10004, 10005 y 10006).
Los supositorios de cada lote fueron almacenados simultáneamente en dos condiciones de almacenamiento: temperatura ambiente (30 ± 2 ºC) y refrigeración (2-8 ºC). El único tipo de envase considerado fue el blíster de aluminio polietileno y la duración del estudio fue de 12 meses. Se siguió la metodología establecida por el Centro Estatal para el Control de Medicamentos, Dispositivos y Equipos Médicos (CECMED),11muestreando individualmente por lote almacenado en cada condición de almacenamiento, al cabo de 1, 3, 6 y 12 meses.
ESTABILIDAD FÍSICA
Se evaluaron las características organolépticas, el peso y el tiempo de liquefacción de los supositorios. En todos los casos los análisis se realizaron por triplicado en cada muestreo (dosis, lote, condición de almacenamiento), siguiendo los procedimientos descritos a continuación:
-Propiedades organolépticas: el muestreo se realizó según se establece en el plan de muestreo por atributo para severidad normal, nivel de inspección II y nivel de calidad aceptable (NCA)= 1. Se evaluaron sensorialmente los supositorios teniendo en cuenta la presencia de brillo, rajaduras, cavidad axial y despuntado, definiendo la aceptación o rechazo del lote según el resultado para cantidad de defectos (d) y su relación con los criterios de aceptación.14
Se consideraron defectos críticos por lo que el valor de NCA fue de 1. Para la inspección normal, nivel de inspección II, muestreo simple y tamaño de muestra n = 20, los valores de aceptación (A= 0) y rechazo (R= 1) fueron los definidos para los criterios de aceptación: si la cantidad de defectos "d"£ A, se acepta, si "d"³ R, se rechaza.
- Determinación del peso promedio: se pesaron 10 supositorios individualmente en balanza analítica SARTORIUS. Luego se sumaron los valores y se dividieron por la cantidad de supositorios pesados para obtener el peso promedio (X). Además se determinó la desviación estándar (DE) y coeficiente de variación (CV).
- Determinación del tiempo de liquefacción: se introdujo el supositorio en una pequeña espiral cónica de alambre inerte, colocándose el conjunto así formado, dentro de un recipiente cilíndrico de vidrio, de aproximadamente 40 mm de diámetro y 80 mL de agua previamente calentada a 37 ± 0,1 ºC y que a su vez se encontraba sumergido en un baño termostatado BM,02 con agitación, a la misma temperatura, debiendo quedar el supositorio 1 o 2 cm por debajo del nivel del agua del recipiente. Se realizaron tres réplicas de este análisis para cada lote. Para fijar el tiempo se tomó como referencia el momento en que se detectó la fusión total de la base al introducir un alambre inerte en la masa del supositorio, procedimiento que se repitió cada 30 s.
ESTABILIDAD QUÍMICA
Se llevó a cabo la determinación del contenido de naproxeno en los supositorios empleando dos métodos analíticos:
- Volumetría de neutralización semiacuosa directa: se utilizó el método volumétrico propuesto en la Farmacopea Británica (BP) del 20112 para el análisis de ingrediente farmacéutico activo (IFA), el cual fue adaptado y validado para el control de calidad15 y los estudios de estabilidad del naproxeno en los supositorios.16 Se aplicó por triplicado a cada lote y los resultados de las réplicas se procesaron para estimar la media, la DE y el CV.
- HPLC: se utilizó el mismo método descrito en la BP 20112 para la cuantificación de naproxeno en supositorios, validado para los estudios de estabilidad.16 Para la cuantificación, se empleó el método de comparación contra patrón, empleando una solución de referencia de naproxeno en fase móvil de igual concentración que la muestra. En cada caso los análisis se realizaron por triplicado. Los análisis fueron solicitados al Centro de Investigación y Desarrollo de Medicamentos (CIDEM). Los resultados fueron procesados por el programa CROMAGATE, obteniendo por cada lote, temperatura de almacenamiento y tiempo de muestreo el contenido de naproxeno en el supositorio (%) y los cromatogramas correspondientes. Las determinaciones solo se realizaron a los 0, 6 y 12 meses para el estudio de vida útil.
Se empleó el método por cromatografía en capa delgada (CCD) validado para estos propósitos17 teniendo en cuenta los parámetros mínimos exigidos para la categoría II prueba límite.5
Criterio de aceptación: El resultado es positivo si se observa la mancha correspondiente al naproxeno bien definida, separada del punto de aplicación y del frente de disolvente con igual valor de Rf e intensidad que el patrón. Se utilizó adicionalmente un patrón de naproxeno equivalente al 90 % cuya intensidad en la mancha siempre debe ser menor a la observada en la muestra, para considerar que la degradación del naproxeno no supera el 10 %. Se utilizó el Rf como criterio de identificación para la presencia de productos de degradación informados con anterioridad.16
PROCESAMIENTO ESTADÍSTICO DE LOS RESULTADOS
Los resultados del estudio de estabilidad se graficaron empleando el programa Excel. Para cada parámetro analizado se construyeron tablas y gráficos de líneas de respuesta obtenida vs. tiempo. Se consideró que "tiempo" equivale al momento del muestreo expresado en "meses". En cada caso los resultados se sometieron a análisis de varianza, con el objetivo de evaluar si existían diferencias significativas entre réplicas y entre lotes. El procesamiento de los resultados se realizó empleando el programa STATGRAPHICS Plus 5.1, opción "comparación", "comparación de varias muestras". La prueba F (tabla ANOVA) permite afirmar que, si p³ 0,05, no existen diferencias estadísticamente significativas para el 95 % de confianza entre los resultados comparados.
Además se realizó la comparación de los resultados del contenido de naproxeno obtenidos por los dos métodos analíticos: volumetría y HPLC, aplicando el mismo procesamiento estadístico descrito anteriormente.
RESULTADOS
Se seleccionaron muestras de 20 unidades para la evaluación de las características organolépticas, en correspondencia con lo establecido en el plan de muestreo según el tamaño del lote y el NCA preestablecido. Los resultados fueron satisfactorios ya que ningún lote fue rechazado según los criterios de aceptación: los supositorios fueron de color homogéneo, con brillo y buena apariencia, sin alteraciones en la superficie.
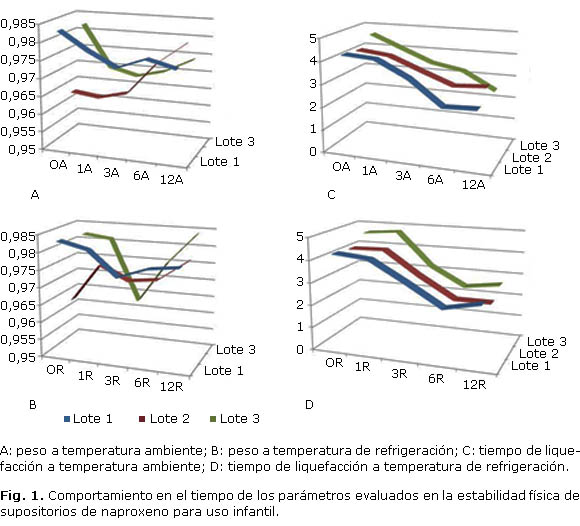
En las figuras 1 y 2 se presentan los resultados obtenidos en las evaluaciones de la estabilidad física, realizadas a los supositorios de naproxeno de cada dosis.
Como se puede apreciar se cumplieron satisfactoriamente los criterios establecidos de peso igual a 1,000 g ± 5 % para los de uso infantil y de 2,000 ± 0,1 g para los supositorios de adultos. Todos los resultados de peso fueron procesados estadísticamente por análisis de varianza de clasificación doble y los resultados no dieron diferencias ni entre lotes, ni entre los diferentes muestreos realizados en cada uno de los estudios.
Los supositorios presentaron tiempos de liquefacción inferiores a los 15 min en todos los muestreos realizados, siendo inferiores para la presentación infantil.
El tiempo de liquefacción mostró una tendencia marcada a la disminución en todos los casos. Desde el punto de vista estadístico estas diferencias se reflejaron fundamentalmente entre las réplicas, es decir, los resultados obtenidos a través de los muestreos realizados en el tiempo, en ambas condiciones de almacenamiento:
- Para supositorios de uso infantil:
- Entre réplicas a 30 ± 2 °C: F calculada= 24,214
- Entre réplicas a 2-8 °C: F calculada= 111, 346
- Para supositorios adulto:
- Entre réplicas a 30 ± 2 °C: F calculada= 19,112
- Entre réplicas a 2-8 °C: F calculada= 36,510
Todas estas diferencias fueron significativas ya que F calculada> F tabulada (5,05).
Además se obtuvieron diferencias entre los lotes solo para el caso de los supositorios para uso infantil almacenados a temperatura de refrigeración (F calculada= 5,714). No obstante, esta pequeña diferencia puede relacionarse con el método por el cual se estimó este parámetro, sujeto a diversas manipulaciones.
Las tablas 1 y 2 resumen los resultados promedios del contenido de naproxeno en los supositorios de cada dosis, así como la identificación de los posibles productos de degradación por CCD expresado por los valores del Rf.


Con relación a la dosis, se obtuvo un mejor comportamiento para los supositorios de menor contenido de naproxeno (50 mg), ya que en los supositorios para adultos (500 mg), los valores superaron el 100 % establecido como valor nominal al realizar la cuantificación por volumetría e incluso por HPLC. No se identificaron productos de degradación por CCD, ya que solo se observó una mancha de igual Rf al naproxeno sustancia de referencia. 17 Estos resultados fueron corroborados con el estudio por HPLC.
En los análisis por HPLC, se obtuvo una señal a tr= 8,724 min, correspondiente al analito y al inicio del cromatograma, se detectaron otras tres señales muy pequeñas atribuibles a las sustancias relacionadas. Este mismo comportamiento se obtuvo durante todo el estudio, independientemente de la temperatura de almacenamiento y del lote estudiado, para los supositorios de ambas dosis almacenados durante 12 meses. A su vez, estos resultados se correspondieron con los obtenidos en el análisis cuantitativo (tablas 1 y 2).
Al realizar el procesamiento estadístico por análisis de varianza, se compararon los % promedios de naproxeno estimados por el método volumétrico y por HPLC. El análisis tuvo en cuenta la posibilidad de diferencias entre lotes y entre réplicas, siendo las réplicas los valores obtenidos a los 0, 3, 6 y 12 meses para cada temperatura de almacenamiento en el caso de la estabilidad acelerada y la vida de estante (solo por volumetría), mientras que para la vida útil en HPLC, se consideraron los datos de 0, 6 y 12 meses. No se obtuvieron diferencias significativas entre lotes, con excepción de los resultados del contenido de naproxeno por HPLC para supositorios de adultos (F calculada para 30 ± 2 °C= 37,875; F calculada para 2-8 °C= 16,981).
En los supositorios para uso infantil se obtuvieron diferencias estadísticamente significativas para los resultados por HPLC entre lotes (F calculada para 30 ± 2 °C= 10,593) y entre réplicas (F calculada para 30 ± 2 °C= 17,185; F calculada para 2-8 °C= 14,852). Sin embargo, en ninguna condición de almacenamiento evaluada se detectaron diferencias para los resultados obtenidos por volumetría. El método indicador de estabilidad, HPLC, fue capaz de detectar los pequeños cambios de concentración de naproxeno en los supositorios de ambas dosis.
Independientemente de las diferencias encontradas entre réplicas por HPLC métodos, la reducción del contenido de naproxeno no puede considerarse un cambio significativo, según la Regulación 23; CECMED.11 Esto significa que desde el punto de vista químico, los supositorios de naproxeno para uso infantil y adulto fueron estables, lo cual se corroboró con la ausencia de productos de degradación. Ninguna de las técnicas cromatográficas aplicadas permitió identificar productos de degradación. Por CCD solo se observó una mancha de igual intensidad y Rf que el naproxeno sustancia de referencia que corresponde con los resultados anteriores17 y por HPLC no se visualizaron incrementos en las señales secundarias atribuibles a sustancias relacionadas, las cuales también estuvieron presentes en la sustancia de referencia.
Por último se muestra la comparación entre los resultados obtenidos por ambos métodos en la tabla 3.
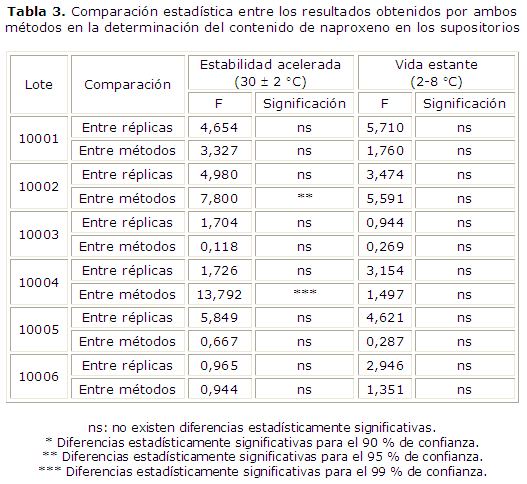
Solo se obtuvieron diferencias estadísticamente significativas para los resultados obtenidos entre métodos para el lote 10002 y el 10004 en la estabilidad acelerada.
En todas las comparaciones realizadas los resultados entre los lotes de la misma dosis, almacenados a la misma temperatura, no fueron diferentes estadísticamente. Estos resultados confirman la gran similitud entre los lotes, ya demostrada desde el punto de vista físico, en este caso, desde el punto de vista químico.
DISCUSIÓN
En los supositorios para uso infantil el 5 % equivale a 0,05 g, por lo que los valores de peso deben estar comprendidos entre 0,95 y 1,05 g. Para los supositorios adultos el peso debe estar comprendido entre 1,900 y 2,100 g. Ni los valores individuales ni los promedios se encontraron fuera de rango durante el estudio realizado, lo cual avala la calidad de los supositorios y la uniformidad del peso en los lotes.5
El comportamiento observado para el parámetro peso demuestra la consistencia del proceso de moldeado, ya que existe una gran similitud de este parámetro entre lotes y fue muy estable en el tiempo. Es decir, el factor temperatura no provocó cambios en el peso de los supositorios.
La relación esperada entre dosis y tiempo de liquefacción, sería un incremento del tiempo a mayor concentración de IFA. A concentraciones mayores de principio activo, existirá mayor dificultad para que ocurra transferencia de calor hacia el interior de la masa del supositorio, que a su vez determina la fusión de este, lo cual fue corroborado según se muestra en los resultados obtenidos.
Los menores tiempos de liquefacción para la presentación infantil se atribuyen al menor diámetro de estos supositorios, lo cual genera una mayor área de contacto con el agua a 37 °C. Este mayor contacto con la temperatura que asemeja la temperatura corporal, conlleva a la liquefacción del supositorio en menor tiempo.
Debido a dificultades en la observación visual de la liquefacción del supositorio, se empleó un procedimiento de ensayo que incluyó el uso de una aguja de alambre inerte que se introdujo cada 30 s en el supositorio. Esto aunque permitió determinar el tiempo en que el excipiente del supositorio se funde, independientemente del grado de desagregación que presentaba, minimiza la influencia del grado de estructuración del supositorio sobre los resultados. No obstante, ante la carencia de un equipo para la realización de este ensayo, el procedimiento aplicado coincide con la metodología estandarizada a nivel nacional por el único laboratorio productor de supositorios.
El comportamiento del parámetro tiempo de liquefacción, pudiera relacionarse con la mayor solubilidad del naproxeno en la base2 durante el tiempo de almacenamiento. Aunque se trata de una suspensión, en ambas dosis queda una parte del naproxeno parcialmente disuelto en la base. Esta disolución no se limita al contacto entre naproxeno y base durante el proceso de elaboración de los supositorios. A medida que transcurre el tiempo, pudiera ocurrir la disolución parcial del naproxeno en la base, ya que se trata de una mezcla de triglicéridos semisintéticos que aunque tiene una apariencia sólida, no es una sustancia pura, sino una mezcla de cristales con líquido interpenetrado con el cual puede interactuar el fármaco. Este fenómeno pudiera ocurrir de forma similar en los supositorios de ambas dosis, ya que se trata de suspensiones del fármaco en grasa saturada.
Otro fenómeno con el cual se pudiera relacionar este comportamiento es el polimorfismo de las bases. Aunque predomina la forma polimórfica más estable, puede existir interacción entre naproxeno y base, aun en estado sólido.
Adicionalmente se encuentra la incidencia de los errores experimentales asociados a este método, los cuales se hacen mayores para variaciones pequeñas de temperatura y otros factores que afectan el mantenimiento de la temperatura constante durante la determinación.
Teniendo en cuenta los resultados de la estabilidad física, se propone el almacenamiento de los supositorios de naproxeno en cualquiera de las temperaturas estudiadas.
Los resultados analíticos obtenidos por los métodos cuantitativos, avalan la estabilidad química de los supositorios de naproxeno. Las diferencias observadas en la comparación entre los métodos se atribuyen a las ventajas del método por HPLC desde el punto de vista de especificidad y sensibilidad, que lo hacen superior a la volumetría para llevar a cabo este tipo de estudio. Aunque durante la etapa de validación se demostró la validez del método volumétrico para el seguimiento de la estabilidad química del naproxeno en supositorios,16 es indiscutible que al ser un método clásico y trabajar en la macroescala, está en desventaja respecto al HPLC y no puede ser considerado como un método indicador de estabilidad. Su uso está limitado como alternativa, ante dificultades de disponibilidad del equipo de HPLC y debe siempre aplicarse conjuntamente con otro método cromatográfico, por ejemplo: CCD.
A partir de los resultados de este trabajo se puede concluir que los supositorios de ambas dosis presentaron elevada estabilidad física y química, independientemente de la temperatura de almacenamiento, la dosis y el lote, durante 12 meses a temperatura ambiente.
Se recomienda extender el estudio para definir el período de validez y la condición de almacenamiento para la comercialización del producto e incluir los estudios de la estabilidad microbiológica correspondientes.
REFERENCIAS BIBLIOGRÁFICAS
1. Ansel HC, Popovich NG, Allen LV. Pharmaceutical Dosage Forms and Drugs Delivery Systems. 6th ed. Baltimore: Lippincott Williams & Wilkins; 1999. p. 106-16.
2. British Pharmacopoeia. Volume I & II Monographs: Medicinal and Pharmaceutical Substances. Naproxen. Naproxen Suppositories [CD-ROM]. London: The Stationery Office; 2011.
3. Clínica Universidad de Navarra. Antiinflamatorios no esteroideos vía sistémica. Naproxeno. [citado 3 Jul 2009]. Disponible en: http://www.cun.es/enfermedades-tratamientos/medicamentos/naproxeno
4. PLM. Diccionario de Especialidades Farmacéuticas [CD-ROM]. 36ta ed. Medellín: Thomson Healthcare; 2008.
5. USP 30. United States Pharmacopoeia XXX and National Formulary 25th [CD-ROM]. United States Pharmacopeial Convention. New York: Pharm Convention Inc.; 2007.
6. Vademecum Internacional [CD-ROM]. 46th ed. 2003 Madrid: Medicom, SA; 2005.
7. Ministerio de Salud Pública. Centro para el Desarrollo de la Farmacoepidemiología. Formulario Nacional de Medicamentos. Cap. 2. La Habana: Editorial Ciencias Médicas; 2006. p. 30, 33.
8. Insel PA. Analgésicos-Antipiréticos, Antiinflamatorios y Fármacos Antigotosos. En: Goodman LS & Gilman A. Las bases farmacológicas de la terapéutica. 9na ed. México, DF: Mc GrawHill-Interamericana; 2002. p. 661-705.
9. Regulación No 16-2006. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Directrices sobre buenas prácticas de fabricación de productos farmacéuticos. Ministerio de Salud Pública. La Habana, Cuba. 2006.
10. ICH QF1. Harmonised Tripartite Guideline. Stability data package for registration applications in climatic zones III and IV. Recommended for Adoption at Step 4 of the ICH Process on 6 February 2003 by the ICH Steering Committee; 2003. [cited 2009 Jul 3]. Disponible en: http://www.emea.eu.int
11. Regulación No 23-2000. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Requerimientos de los estudios de estabilidad para el registro de productos farmacéuticos nuevos y conocidos. Ministerio de Salud Pública. La Habana, Cuba. 2000.
12.Regulación No 24-2000. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Requerimientos de los estudios de estabilidad para el registro de nuevos ingredientes activos farmacéutico. Ministerio de Salud Pública. La Habana, Cuba. 2000.
13. No 25-2000. Centro para el Control Estatal de la Calidad de los Medicamentos (CECMED). Requerimientos de los estudios de estabilidad para el registro de productos biológicos y biotecnológicos. Ministerio de salud pública. La Habana, Cuba. 2000.
14. Espinosa JM. Normalización, Metrología y Control de la Calidad. La Habana: Ed. Félix Varela; 2008. p. 152-60.
15. Rodríguez Hernández Y, Suárez Pérez Y, García Pulpeiro O, Hernández Contreras OY. Validación de métodos analíticos para el control de calidad de Naproxeno supositorios. Rev Cubana Farm. 2011;45(3):341-54.
16. Rodríguez Hernández Y, Suárez Pérez Y, García Pulpeiro O, Rodríguez Borges T, Alonso H. Validación de métodos analíticos para los estudios de estabilidad del Naproxeno en supositorios para uso infantil y adulto. Rev Cubana Farm. 2011;45(4):494-504.
17. Rodríguez Hernández Y, Suárez Pérez Y, García Pulpeiro O, Rodríguez Borges T. Desarrollo y validación de un método por cromatografía en capa delgada para los estudios de estabilidad del Naproxeno supositorios. Rev Cubana Farm. 2011;45(4):480-93.
Recibido: 19 de diciembre de 2013.
Aprobado: 20 de enero de 2014.
Yania Suárez Pérez . Instituto de Farmacia y Alimentos (IFAL). Ave 23 No. 21425 e/ 214 y 222, La Coronela, La Lisa. La Habana, Cuba. Correo electrónico: yaniasp@ifal.uh.cu

{kind=link}
{kind=link}