










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr vol.87 no.2 Ciudad de la Habana abr.-jun. 2015
PRESENTACIÓN DE CASO
Síndrome de Ondina, hipoventilación central congénita idiopática
Ondine´s curse or idiopathic congenital central hyperventilation syndrome
Daniel Jiménez Marín, Ana María Londoño Salinas, Sara Rojas Jiménez, Johan Sebastián Lopera Valle
Internos de Medicina. Facultad de Medicina. Escuela de Ciencias de la Salud. Universidad Pontificia Bolivariana. Medellín, Colombia.
RESUMEN
El síndrome de hipoventilación central congénito idiopático, también conocido como síndrome de Ondina, es una enfermedad poco común, caracterizada por la ausencia congénita del control central de la respiración y disfunción del sistema nervioso autónomo. Su incidencia se estima en aproximadamente 1 de cada 200 000 nacimientos al año. Es una enfermedad de transmisión autosómica dominante, derivada de una mutación heterocigótica del gen PHOX2B, presente en 90 % de los pacientes, pero su causa fisiopatológica aún no está claramente dilucidada. Presenta una elevada tasa de mortalidad, y una dependencia a la ventilación mecánica durante el sueño de por vida; sin embargo, gracias a una atención multidisciplinar y coordinada, con estrecha vigilancia y apoyo, podría esperarse que los pacientes con ella puedan llevar una vida relativamente normal. Se realiza el reporte de un caso diagnosticado en el departamento de Antioquia (Colombia), lugar con importantes barreras socioeconómicas que limitan el estudio complejo de este tipo de enfermedades de baja prevalencia global.
Palabras clave: apnea del sueño central, polisomnografía, respiración artificial, mutación.
ABSTRACT
Idiopathic congenital central hypoventilation syndrome, also known as Ondine´s curse, is a rare disease characterized by congenital absence of the central control of breathing and autonomic nervous system dysfunction. Its incidence is estimated to be one per 200,000 births a year. It is an autosomal dominant disease derived from a heterozygous PHOX2B gene mutation, present in 90 % of patients, but its pathophysiological cause has not been elucidated yet. It has a high mortality rate and a lifelong dependency on a life-support device during sleep. However, it could be expected that a coordinated multidisciplinary care, with close monitoring and support, could help this kind of patients to have a relatively normal life. This is the report of a case diagnosed with this disease in Antioquia, Colombia, a place with significant socio-economic limitations that hinder a complete study of this type of low overall prevalence disease.
Keywords: central sleep apnea, polysomnography, artificial respiration, mutation.
INTRODUCCIÓN
El síndrome de hipoventilación central congénito idiopático (SHCCI), conocido también con los nombres de hipoventilación alveolar primaria y síndrome o maldición de Ondina, es un desorden genético, poco frecuente, caracterizado por hipoventilación alveolar y una alteración del control autónomo de la ventilación, en ausencia de enfermedad primaria que lo justifique.1 Su prevalencia es baja, se estima 1 caso por cada 200 000 nacidos vivos,2 y han sido reportados en la literatura médica internacional alrededor de 1 000 niños con el síndrome, aunque esta cifra puede estar subestimada.3
A continuación se realiza el reporte de un caso diagnosticado en el departamento de Antioquia (Colombia), con el fin de dar a conocer su presentación clínica en este medio, el cual tiene importantes barreras socioeconómicas que limitan el estudio complejo de este tipo de enfermedades de baja prevalencia global.
CASO CLÍNICO
Paciente masculino, de 22 días de vida, hospitalizado en una institución de alta complejidad para estudio de ictericia neonatal. Tras 2 días de estancia hospitalaria, presenta falla ventilatoria por períodos de bradipnea y apnea, y es trasladado a la unidad de cuidados intensivos neonatales, con gases arteriales compatibles con alcalosis respiratoria. El paciente se torna dependiente del ventilador, tolera extubación por algunas horas, para posteriormente presentar compromiso del estado general, hipotonía, palidez, poco esfuerzo respiratorio, respiración superficial y períodos de apnea. Adicionalmente presenta acidosis respiratoria severa con retención de CO2, y en ocasiones, crisis convulsivas, por lo que fue imperiosa la necesidad de reintubación en múltiples ocasiones.
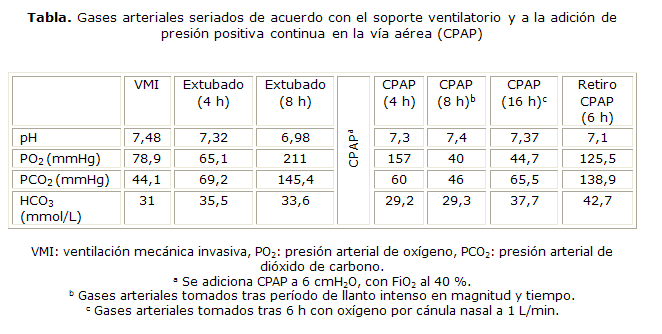
El paciente no tolera la disminución en los parámetros ventilatorios ante la escasa respuesta respiratoria espontánea, la cual no se asocia a obstrucción de la vía aérea superior ni compromiso de la inferior. Se implementa presión positiva continua en la vía aérea (CPAP) en los periodos de sueño, y se realizan gases arteriales seriados cada 4 horas (tabla).
Se descarta error congénito del metabolismo, además de otras causas de hipoventilación central, miopatías y neuropatías, razones por las cuales se sospecha diagnóstico de síndrome de Ondina. Al mes y 3 días de vida se realiza polisomnograma con video sincronizado, y se administra presión positiva bifásica de la vía aérea (BIPAP) durante la primera mitad de la noche (presión positiva inspiratoria [IPAP] de 16 cmH2O, y espiratoria [EPAP] de 4 cmH2O), y retiro de este durante la segunda mitad.
En esta prueba se encuentra una saturación de oxihemoglobina promedio de 94 %, que permanece estable con o sin BIPAP. El nivel de CO2 fue mayor de 45 mmHg únicamente en el 5 % del tiempo con BIPAP, mientras que en la segunda mitad de la noche (sin BIPAP), se mantuvo por encima de 46 mmHg, con un valor máximo de 64 mmHg, y con cifras menores a 45 mmHg solo cuando el paciente despertaba. Se concluye finalmente la compatibilidad del polisomnograma con el diagnóstico de síndrome de hipoventilación central asociada al sueño.
Igualmente, se realiza electroencefalograma (EEG), que muestra trazado electroencefalográfico de paciente en estado de sueño No MOR 1 y 2 con hallazgos anormales por actividad epileptiforme temporoparietal izquierda, asociado a disminución de los grafoelementos del sueño en el hemisferio izquierdo. No se registraron crisis clínicas. En la figura se observa EEG de control realizado a la semana, en el cual no se evidencian las anomalías previamente referidas.
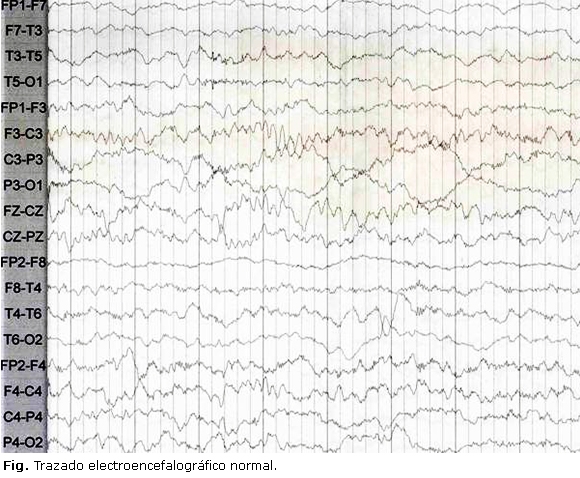
Posteriormente, se traslada al servicio de cuidados intermedios, luego de haber logrado la extubación y tratamiento exitoso con BIPAP durante el sueño, garantizando siempre una saturación mayor del 92 %. Se da de alta a los 6 meses de vida, tras un total de 4 meses de estancia hospitalaria en unidad de cuidados intensivos y un mes más en unidad de cuidados intermedios. Se dan órdenes para terapia respiratoria y física interdiaria, BIPAP 16/4 a 0,5-1 L/min durante el sueño, y cita ambulatoria de control en 6 semanas con Neuropediatría.
DISCUSIÓN
El SHCCI es producto de un defecto primario del control autonómico de la respiración. A pesar de que el mecanismo exacto de la enfermedad es aún desconocido, se considera que un trastorno en la integración de la información proveniente de los quimiorreceptores, probablemente a nivel del tronco encefálico, podría explicarla.4 Por ello, la expresión clínica es más evidente ante la ausencia o menor actividad de los centros de control respiratorio involuntario durante el sueño, especialmente durante la fase de ausencia de movimientos oculares rápidos (No MOR).5
En el año 2003 se identificaron mutaciones en el gen PHOX2B ubicado en el cromosoma 4 (4p12) como responsables de la génesis del SHCCI, y se determinó, igualmente, su patrón de herencia como autosómico dominante.1,6,7 Este gen está relacionado con la migración y diferenciación de células de la cresta neural, por lo que en pacientes SHCCI pueden evidenciarse síntomas de disfunción del sistema nervioso autónomo (SNA).8,9
No existe ningún signo clínico patognomónico del síndrome; su presentación clásica es en el recién nacido, donde se encuentran frecuencias respiratorias monótonas y respiración superficial durante el sueño, o con alteración en vigilia en casos más severos. Con frecuencia se asocian hipotonía muscular en diversos grados y signos de disfunción autonómica.9 En ciertos casos el inicio de las manifestaciones puede ser más tardío, e incluso, menos severo, con cuadros de cianosis, edema, taquicardia, diaforesis y signos de insuficiencia cardíaca derecha durante el sueño.1,10 Así mismo, algunos pacientes pueden iniciar con sintomatología más florida y complicaciones que requieren tratamiento exhaustivo. Este grupo conforma la presentación atípica del SHCCI, y puede incluir síntomas de disfunción orgánica por hipoxemia crónica e hipercapnia, enfermedad cardíaca pulmonar (cor pulmonale), convulsiones o retraso en el desarrollo psicomotor.2,11 En el caso de este paciente en particular, los hallazgos clínicos indicativos de afectación ventilatoria fueron evidentes desde la cuarta semana de vida, con episodios de cianosis espontánea, desaturación, y falla ventilatoria progresiva con compromiso multiorgánico, que llevó a requerimientos crecientes en cuanto a medidas terapéuticas.
Para realizar el diagnóstico se recomiendan los criterios propuestos en 2010 por la Sociedad Torácica Americana,3 a partir de los cuales se hace necesario demostrar la presencia de hipoventilación con respuesta ventilatoria ausente o atenuada a la hipercapnia y/o hipoxemia; hipoventilación con frecuencia respiratoria normal y volumen corriente disminuido únicamente durante el sueño, con compromiso en vigilia igualmente; ausencia de percepción de asfixia con desarrollo de compromiso fisiológico secundario a la hipercapnia y/o hipoxemia; presencia de síntomas de disfunción autonómica; y finalmente, la ausencia de enfermedad primaria, que pudiera ser responsable de toda la constelación de signos y síntomas.3
Es igualmente necesaria la realización de una prueba de pesquisa para mutaciones en el gen PHOX2B;10,12 sin embargo, es aún un aspecto que no es posible abarcar en todos los casos, principalmente en países en vía de desarrollo como Colombia.13 Es debido a las limitaciones para el acceso a estas pruebas que se debe justificar el diagnóstico con base en los criterios clínicos antes expuestos, y la exclusión de otras enfermedades compatibles, y es de vital importancia la realización de una polisomnografía, la cual se postula como un método preciso para la evaluación de la fisiología pulmonar con base en el ciclo sueño-vigilia.14
El objetivo clave del tratamiento es asegurar una ventilación adecuada durante la vigilia y el sueño, para lo cual existe una variedad de modalidades de ventilación, además de la estimulación diafragmática con un marcapasos en mayores de 5 años.1,11 El seguimiento clínico periódico de las personas afectadas es obligatorio, y es indispensable un acceso multidisciplinario para proporcionar un tratamiento integral y detectar oportunamente las complicaciones.11
Las particularidades del caso presentado llevan a establecer el diagnóstico de SHCCI, por presentar una falla del control central de la respiración sin otra causa demostrable que lo explique. Es igualmente importante examinar en todo caso la presencia de neurocrestopatías y alteraciones autonómicas, en el paciente y en su familia, además de instaurar vigilancia de expresión tardía de la enfermedad en sus familiares.8 Por último, en este caso sería de bastante utilidad la realización de un estudio genético, con el fin de determinar la mutación específica y su posible relación con el fenotipo expresado.11,15
En conclusión, el SHCCI es una enfermedad que constituye, sin duda alguna, un reto diagnóstico, debido a la variabilidad en la presentación clínica y a las limitaciones en cuanto al completo acceso a las pruebas diagnósticas, especialmente las genéticas, en países con barreras socioeconómicas y políticas importantes como Colombia. Indudablemente el tratamiento a estos pacientes debe estar a cargo de un vasto grupo de especialistas organizados en un equipo multidisciplinario, el cual brinde el adecuado apoyo social y psicológico necesario para el paciente y su familia, ya que, con un atento seguimiento, los niños afectados pueden obtener mayor esperanza y calidad de vida.
REFERENCIAS BIBLIOGRÁFICAS
1. Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, et al. Idiopathic congenital central hypoventilation syndrome: Analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2B. Am J Med Genet A. 2003;123A:267-78.
2. Trang H, Dehan M, Beaufils F, Zaccaria I, Amiel J, Gaultier C; French CCHS Working Group. The French Congenital Central Hypoventilation Syndrome Registry: general data, phenotype, and genotype. Chest. 2005;127(1):72-9.
3. Weese-Mayer DE, Berry-Kravis EM, Ceccherini I, Keens TG, Loghmanee DA, Trang H. Congenital central hypoventilation syndrome: genetic basis, diagnosis, and management. Am J Respir Crit Care Med. 2010;181:626-44.
4. Crowell BA, Bissinger RL, Conway-Orgel M. Congenital central hypoventilation syndrome: a case report. Adv Neonatal Care. 2011;11(3):167-72.
5. Healy F, Marcus CL. Congenital Central Hypoventilation Syndrome in Children. Paediatr Respir Rev. 2011;12(4):253-63.
6. Amiel J, Laudier B, Attie ìBitach T, Trang H, de Pontual L, Gener B, et al. Polyalanine expansion and frameshift mutations of the paired-liked homeoboxgene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459-61.
7. Meguro T, Yoshida Y, Hayashi M, Toyota K, Otagiri T, Mochizuki N, et al. Inheritance of polyalanine expansion mutation of PHOX2B in congenital central hypoventilation syndrome. J Hum Genet. 2012;57(5):335-7.
8. Lee CW, Lee JH, Jung EY, Choi SO, Kim CS, Lee SL, Kim DK. Haddad syndrome with PHOX2B gene mutation in a Korean infant. J Korean Med Sci. 2011;26(2):312-5.
9. Ramanantsoa N, Gallego J. Congenital central hypoventilation syndrome. Respir Physiol Neurobiol. 2013;189(2):272-9.
10. Loghmanee DA, Rand CM, Zhou L, Berry-Kravis EM, Jennings LJ, Yu M, et al. Paired-like homeobox gene 2b (PHOX2B) and congenital central hypoventilation syndrome (CCHS): genotype/phenotype correlation in cohort of 347 cases. Am J Respir Crit Care Med. 2009;179:A6341.
11. Joseph L, Goldberg S, Shahroor S, Gomori M, Mimouni FB, Picard E. Sinus vein thrombosis as presenting finding in the congenital central hypoventilation syndrome: an insight on the pathophysiology of the association. Pediatr Pulmonol. 2011;46(8):826-8.
12. Jennings LJ, Yu M, Rand CM, Kravis N, Berry-Kravis EM, Patwari PP, et al. Variable human phenotype associated with novel deletions of the PHOX2B gene. Pediatr Pulmonol. 2012;47(2):153-61.
13. Alarcón J, Rojas JP, Meneses DK, Ocampo GM, Patiño P. Síndrome de hipoventilación central. Acta Neurol Colomb. 2013;29(3):209-14.
14. Urquhart D. Investigation and management of childhood sleep apnoea. Hippokratia. 2013;17(3):196-202.
15. Trivedi A, Waters K, Suresh S, Nair R. Congenital central hypoventilation syndrome: four families. Sleep Breath. 2011;15(4):785-9.
Recibido: 24 de febrero de 2014.
Aprobado: 23 de septiembre de 2014.
Daniel Jiménez Marín. Facultad de Medicina. Escuela de Ciencias de la Salud. Universidad Pontificia Bolivariana. Calle 78B # 72A-109. Medellín, Colombia. Correo electrónico: loperavalle@hotmail.com


{kind=link}