










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El término ataxia refiere al signo neurológico que indica una alteración de la coordinación motora voluntaria y del control postural.1 Este trastorno es resultado de una disfunción del cerebelo y/o sus vías aferentes y eferentes, así como alteraciones en la médula espinal, nervios periféricos o una combinación de estas tres condiciones.2 Las ataxias pueden clasificarse en esporádicas, hereditarias y adquiridas según las causas que las originan.
Las ataxias hereditarias son las más estudiadas y se clasifican según el patrón de herencia en autosómicas dominantes, autosómicas recesivas, ligadas al cromosoma X y de herencia mitocondrial. Las ataxias autosómicas recesivas más conocidas son la ataxia de Friedreich y la ataxia telangiectasia. Las ataxias autosómicas dominantes se caracterizan por una degeneración del cerebelo, la médula espinal y sus vías de conexión, por lo que son conocidas como ataxias espinocerebelosas.2 De estas ataxias existen 48 formas moleculares hasta la fecha.
Las ataxias esporádicas son aquellas que no se conocen las causas de su aparición. En este grupo se encuentran la atrofia multisistémica tipo cerebelosa, caracterizada por una atrofia del tallo cerebral y afectaciones de los ganglios basales; y la ataxia cerebelosa idiopática de inicio tardío en el adulto, caracterizada por un síndrome cerebeloso de progresión lenta con manifestaciones de ataxia de la marcha y postural, dismetría de miembros superiores, temblores, disartria y trastornos oculomotores.
Por otra parte, las ataxias adquiridas pueden ser resultado de diversas causas. Pueden tener origen tóxico debido a la ingesta de cantidades supraterapeúticas de fármacos anticonvulsivantes, antihistamínicos, antidepresivos, consumo de alcohol e inhalación de monóxido de carbono. También pueden tener un origen traumático, que forma parte del síndrome posconcusión o luego de un trauma cervical por disección de la arteria vertebral. También se incluyen las causadas por tumoraciones en el tallo encefálico o en el cerebelo y por déficit de vitaminas, principalmente de vitamina B12. Otras de las causas más comunes son los accidentes vasculares de tipo hemorrágicos o isquémicos, principalmente aquellos que comprometen a estructuras cerebelosas.
En las ataxias adquiridas se incluyen las ataxias de origen infeccioso, causadas por la infección por bacterias, parásitos, hongos o virus neurotrópicos. En la literatura se informan diversos casos de ataxias secundarias provocadas por estos virus. Dentro de los casos más descritos se encuentran la ataxia cerebelosa aguda, la ataxia aguda posinfecciosa, el síndrome opsoclono-mioclono-atáxico (SOMA) y la ataxia por encefalomielitis aguda diseminada. Sin embargo, de este tipo de ataxias se conoce poco y aún no está descrito su mecanismo neuropatogénico. Por tanto, el objetivo de este trabajo es actualizar los conocimientos relacionados con las ataxias causadas por virus neurotrópicos y los mecanismos neurodegenerativos que pudieran tener relación con este trastorno del movimiento.
Métodos
Se realizó una revisión bibliográfica de artículos publicados en las principales bases de datos bibliográficas (Web of Sciences, SCOPUS, SciELO, PubMed). Se revisaron un total de 79 artículos comprendidos en los últimos 26 años, de los cuales 17 son revisiones bibliográficas, 29 artículos originales, 25 reportes de casos, dos artículos de opinión, tres cartas al editor y tres libros. Se excluyeron aquellos artículos que no fueron publicados en los últimos 26 años. Para la búsqueda se utilizaron las palabras claves: ataxia, virus neurotrópicos, ataxias secundarias, ataxias infecciosas, ataxias cerebelosas, en inglés y español.
Análisis e integración de la información
Virus neurotrópicos
Los virus neurotrópicos son aquellos que presentan gran afinidad y capacidad de invadir, infectar o persistir en el tejido nervioso, ya sea del sistema nervioso central (SNC) y/o del periférico, lo que origina diversos cuadros neurológicos. El mecanismo de acción parece ser la replicación viral directa en este tejido con la subsecuente destrucción neuronal y el desencadenamiento de mecanismos inmunológicos que condicionan la afectación al tejido. Algunos virus neurotrópicos, después de la primoinfección, persisten de forma latente sobre el tejido nervioso, los que se pueden reactivar en situaciones de inmunosupresión.
El mecanismo por el cual estos virus ingresan al SNC no está bien definido. Se proponen dos rutas: la diseminación por transporte axonal, ya sea retrógrado o anterógrado, y la diseminación hematógena (Fig.). En la diseminación por transporte axonal retrógrado, los virus viajan desde los axones hasta el soma neuronal, y se trasladan hacia las neuronas del SNC por las fibras motoras de los nervios periféricos. En el caso anterógrado los virus viajan desde el soma hacia los axones, y se mueven por las fibras sensitivas de los nervios periféricos hasta llegar al SNC (Fig., A). En la diseminación hematógena los virus atraviesan la barrera hematoencefálica (BHE) por tres posibles vías: la transcitosis por células endoteliales infectadas; el paso de la BHE a través del líquido cefalorraquídeo (LCR) y a través de macrófagos y microglias infectados (mecanismo del caballo de Troya) (Fig., B). La entrada de estos virus al SNC desencadena mecanismos de activación microglial3 que conducen a la liberación de quimiocinas y citocinas que causan la muerte neuronal.4
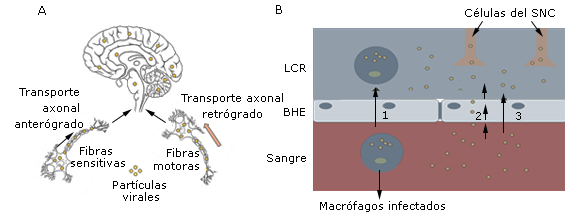 A: Diseminación por transporte axonal anterógrado o retrógrado. B: Diseminación hematógena a través de macrófagos infectados (1), transcitosis por células endoteliales (2) y a través de las hendiduras de la BHE (3).
A: Diseminación por transporte axonal anterógrado o retrógrado. B: Diseminación hematógena a través de macrófagos infectados (1), transcitosis por células endoteliales (2) y a través de las hendiduras de la BHE (3).Fig Rutas utilizadas por los virus neurotrópicos para invadir el SNC.
La apoptosis de células neuronales inducida por virus es una de las principales causas de la neurodegeneración. Este proceso se desencadena como una respuesta al estrés metabólico provocado por la replicación viral en la célula hospedera. Los virus neurotrópicos estimulan el estrés oxidativo y conducen a procesos de estrés mitocondrial y del retículo endoplásmico. La inducción de estrés y el inicio del proceso de neurodegeneración por la infección pueden tener un efecto persistente en el hospedero.4
Se consideran virus neurotrópicos los virus de la familia Herpesviridae, con gran afinidad por el sistema nervioso, entre los que se destacan: herpes virus simple tipo 1 y 2, el virus varicela zóster, Epstein-Barr, citomegalovirus y el herpes humano tipo 6. Se incluyen los arbovirus, como el virus de la encefalitis equina del este, el virus de la encefalitis equina del oeste, el virus de la encefalitis equina venezolana; y también los del género Flavivirus, como el virus del Nilo Occidental, dengue y zika, que pueden condicionar cuadros neurológicos agudos graves. También los virus de la rubeola, hepatitis B, así como influenza A H1N1, pueden generar cuadros de encefalitis, síndrome de Guillain-Barré y su variante Miller-Fisher.5 Otros virus neurotrópicos son los enterovirus, tales como el virus coxsackie, enterovirus 71, poliovirus y los echovirus tipos 6 y 9. Además se hace referencia del neurotropismo al VIH, rotavirus, parvovirus B19, virus JC, adenovirus, entre otros.1 Muchos de estos virus constituyen agentes etiológicos causantes de ataxia. Esto está demostrado mediante la detección directa de partículas virales o de su genoma, así como anticuerpos específicos contra estos virus en el suero o en el LCR de pacientes con ataxias infecciosas.6,7,8,9
Una evidencia que relaciona algunos de estos virus con las enfermedades neurodegenerativas es la presencia de un alto nivel de coincidencia de octapéptidos de los virus hepatitis C, VIH-2, Epstein-Barr, herpes humano tipo 6 y el citomegalovirus con antígenos del cerebro humano, asociados a enfermedades como la esclerosis lateral amiotrófica, ataxia cerebelosa, enfermedad de Huntington, enfermedad de Parkinson, deterioro cognitivo, afasia y apraxia oculomotora. Esta coincidencia peptídica consiste en que muchas secuencias comunes son repeticiones de un solo aminoácido y que en su mayoría, los octapéptidos compartidos son parte de epítopos validados experimentalmente, lo que sugiere una potencial reacción inmune cruzada de los péptidos virales compartidos con los antígenos cerebrales que participan en la neurodegeneración. Este estudio puede tener relevancia en la elaboración de terapias basadas en péptidos que bloqueen las posibles reacciones autoinmunes cruzadas en enfermedades neurológicas.10
Actualmente se desconocen los mecanismos directos que relacionan a estos virus con la aparición de la ataxia. A continuación se describen una serie de ejemplos de virus neurotrópicos que se han detectado en casos de pacientes con ataxias infecciosas (cuadro), y los mecanismos neurodegenerativos que pudieran relacionarse con el origen de estas ataxias.
Cuadro Virus neurotrópicos que causan ataxia
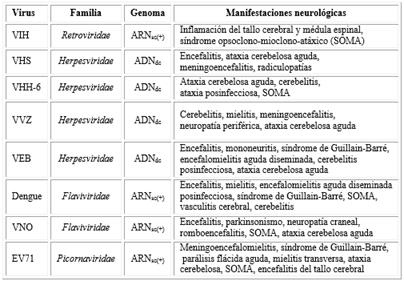
VIH: virus de la inmunodeficiencia humana; VHS: virus del herpes simple; VHH-6: virus del herpes humano tipo 6; VVZ: virus de la varicela zoster; VEB: virus Epstein-Barr; VNO: virus del Nilo Occidental; EV71: enterovirus 71; ARNsc(+): ARN de simple cadena con polaridad positiva; ADNdc: ADN de doble cadena.
Virus de la inmunodeficiencia humana (VIH)
Diversos mecanismos patogénicos directos e indirectos podrían contribuir al daño neuronal causado por la infección por el VIH. Durante las etapas iniciales de la infección, el virus penetra en el cerebro a través de células mieloides y linfoides migratorias y en algunos pacientes infecta los macrófagos perivasculares, microglias y astrocitos.11,12 Algunos de los principales mecanismos patogénicos relacionados con la neurodegeneración son la inflamación crónica del SNC12) y la infección de monocitos y macrófagos del SNC, en que estas células actúan como reservorio viral.13
La infección latente y sostenida de las meninges, y probablemente del cerebro y cerebelo, puede causar una inflamación continua del SNC debido a la toxicidad directa e indirecta de proteínas virales reguladoras y la glicoproteína de la envoltura gp120.14 Estas proteínas activan a macrófagos antiinflamatorios presentes en el LCR a través de citocinas y los convierten en proinflamatorios.15 La expresión crónica de la proteína viral reguladora Tat en ratones se asoció con una reducción del volumen cerebral y daño neuronal,11 a través de la activación del factor de necrosis tumoral alfa, por alteración en la regulación de la homeostasis del calcio celular, excitotoxicidad a través de la activación del receptor del N-metil-D-aspartato (NMDA), hiperpolarización mitocondrial y alteración de la expresión de microARNs.16 También se ha demostrado en ratones, que gp120 provoca pérdida de sinapsis, alteración del crecimiento de las células nerviosas y astrocitosis.17
Los mecanismos fisiopatogénicos exactos de la ataxia no están demostrados, pero parecen estar mediados por mecanismos autoinmunes o por el efecto neurotóxico directo del virus que conduce a la apoptosis de las células de Purkinje y células granulosas.18 Estudios han demostrado una degeneración de la capa celular del cerebelo, así como inflamación axonal en el tallo cerebral y la médula espinal, lo que refuerza la idea de un proceso neurodegenerativo.19 En un estudio, 40 pacientes con infección por VIH fueron evaluados en cuanto a la pérdida de equilibrio, y estos hallazgos se correlacionaron con un deterioro del tracto pontocerebeloso.20
Como una evidencia de mecanismo autoinmune, se detectó en el suero de un paciente infectado con VIH que presentaba manifestaciones atáxicas, la presencia del anticuerpo anti-Yo, encontrado mayoritariamente en el SOMA paraneoplásico, y el anticuerpo antigliadina, asociado con la ataxia por gluten.21 Anti-Yo fija el complemento y reacciona contra antígenos citoplasmáticos de las células de Purkinje de 34 y 62 kDa, lo cual provoca la muerte de estas células.22 Por otra parte, antigliadina reacciona contra la sinapsina I neuronal, fosfoproteína con función enzimática que regula la liberación de neurotransmisores. Esto impide la comunicación neuronal, lo que causa neurodegeneración.23
El SOMA puede ser provocado por la infección por VIH. Este trastorno puede ocurrir durante la infección temprana, por un síndrome de reconstitución inmune o en asociación con otras infecciones, especialmente tuberculosis.24
Herpesvirus. Virus del herpes simple (VHS)
Los herpesvirus tienen alta prevalencia a nivel global, más del 80 % de los adultos se encuentran infectados y por lo general la infección ocurre desde edades tempranas de la vida. Estos virus producen latencia de por vida en el hospedero infectado. Los herpesvirus ingresan a las neuronas sensoriales periféricas en sus terminales y se transportan a través del transporte axonal retrógrado hacia el núcleo, en el que pueden persistir como un episoma circular. La expresión viral está restringida a las transcripciones asociadas a la latencia.
Después de la inoculación periférica, estos virus llegan a los ganglios donde se replican los primeros días después de la infección y establecen la latencia. Se plantea que el control inmune de la replicación juega un papel clave en el establecimiento de la latencia viral, y se debe principalmente a la acción de las células T CD8+. Aunque, las neuronas pueden restringir la expresión de genes inmediatamente después de la entrada del virus, la latencia puede interrumpirse por episodios periódicos de reactivación. Sus factores desencadenantes están relacionados con la señalización neuronal específica, activada por estímulos como el estrés físico o emocional, fiebre, exposición a la luz ultravioleta, menstruación o desequilibrio hormonal. La inmunosupresión también facilita la recurrencia de la infección, pues disminuye el control inmunológico sobre la replicación viral.25,26
Entre los herpesvirus más conocidos está el VHS, el cual presenta dos serotipos que comparten una alta homología en el genoma. La virulencia del VHS se debe principalmente a su capacidad para invadir el SNC desde un sitio periférico de entrada, y luego replicarse en las neuronas del cerebro y cerebelo. Se ha demostrado que el virus se replica en el portal de entrada, luego en los núcleos ganglionares del trigémino, y en el SNC,26 en el que se detectan gliosis, astrocitosis, afectaciones en los oligodendrocitos y proliferación de microglias. Las inclusiones virales intranucleares en las células del SNC son inconstantes, los antígenos virales son detectables solo en la etapa temprana de la enfermedad, aunque el ADN viral permanece detectable en el LCR y en el tejido cerebral de los pacientes infectados.26,27,28
La proteína γ134.5 del virus es el factor clave para la replicación viral en el SNC, pues está involucrada en la inhibición de la respuesta inmune innata y de la autofagia celular.26 Existen evidencias de mecanismos inmunológicos relacionados con las manifestaciones neurológicas. La generación de anticuerpos antirreceptor NMDA, comúnmente presentes en la encefalitis autoinmune en pacientes menores de 30 años, constituye un mecanismo neuropatogénico.27 Este receptor juega un papel importante en la neurotransmisión glutamatérgica que envía señales excitatorias a las neuronas, por lo que estos anticuerpos impiden la comunicación interneuronal, lo cual provoca neurodegeneración.
La infección por el VHS es una de las causas más comunes de encefalitis aguda. Por lo general, se manifiesta como una infección necrotizante aguda y focal que afecta a los lóbulos frontales y temporales del cerebro, y también a los lóbulos parietales y occipital.25 Además puede infectar al cerebelo que causa ataxia de la marcha, nistagmos, dismetrías y dificultad en el habla. Los casos de ataxia cerebelosa causados por la infección por VHS son muy raros. Solamente tres casos han sido informados en la literatura. La frecuencia de la ataxia cerebelosa aguda causada por VHS es de 0,4 % de todas las presentaciones neurológicas pediátricas y la incidencia anual de la cerebelitis aguda es de 5,9 pacientes por cada 100 000.28
Virus del herpes humano tipo 6 (VHH-6)
El VHH-6 es uno de los virus más neurotrópicos. La infección activa causa enfermedades severas del SNC en pacientes adultos inmunocompetentes e inmunocomprometidos, que se caracterizan por ser enfermedades multifocales desmielinizantes.29 Mecanismos como la invasión directa del virus y la respuesta inmune del hospedero están implicados en la patogenia de las complicaciones del SNC. La neuroinvasividad del virus se demuestra por el hecho de que el ADN se detecta con frecuencia en distintas regiones del encéfalo.30 Ambas variantes A y B de VHH-6 se han encontrado en el tejido cerebral, con más frecuencia la presencia de B, aunque A tiene mayor neurotropismo.30
Un mecanismo neurodegenerativo causado directamente es la infección de los oligodendrocitos, células que se encargan de la producción de la vaina de mielina en el SNC. Otro mecanismo, pero de naturaleza autoinmune, también daña a los oligodendrocitos, el mismo está relacionado con la presencia del anticuerpo anti-MOG, el cual reacciona con la glicoproteína de la mielina del oligodendrocito, que causa la desmielinización del SNC.31,32 También existen informes de presencia de antígenos y del ADN del VHH-6 en astrocitos y microglias.33 Además, se refiere que el VHH-6 infecta a monocitos y macrófagos migratorios, lo cual llega hasta el SNC por diseminación hematógena.34 Por otra parte, las interacciones entre la inmunidad del individuo hospedero y las células gliales del SNC después de la infección podrían contribuir a la neuropatogenia del SOMA.35
La infección por VHH-6 se relaciona con la ataxia cerebelosa aguda en niños y con la ataxia posinfecciosa aguda. El primer informe de ataxia causada por VHH-6 fue informado en el 2003 en un caso de roséola infantil,36 y a partir de ese momento se ha descrito como agente etiológico de algunos casos de ataxia aguda en niños con detección del virus en LCR.37 La manifestación más severa es la cerebelitis, caracterizada por síntomas neurológicos agudos y sistémicos asociados a anomalías cerebelosas.38 La ataxia posinfecciosa aguda y el SOMA son alteraciones neurológicas mediadas por el sistema inmune, debido a un proceso inflamatorio en el cerebelo, en que la infección por VHH-6 es una de las causas.35 Este fenómeno autoinmune podría ser desencadenado por la infección, con la posterior producción de anticuerpos que reaccionan de forma cruzada contra epítopos cerebelosos.39
Un estudio de resonancia magnética nuclear (RMN) realizado en una paciente con ataxia aguda infectada con VHH-6, revela la presencia de lesiones en el cerebelo que se extendían a otras regiones del SNC, lo cual provoca una encefalomielitis aguda diseminada,37 en ocasiones esta afectación se relaciona con la ataxia aguda.8,31 En la mayoría de los casos informados los pacientes se han recuperado satisfactoriamente de las manifestaciones cerebelosas con una evolución benigna y sin secuelas.8,35,37 Sin embargo, se han descrito casos más graves, con secuelas neurológicas, probablemente por afectación del parénquima cerebral.40 Las secuelas son más propensas a desarrollarse si la infección involucra también al cerebro, además del cerebelo.37
Virus de la varicela zoster (VVZ)
El VVZ es el agente causal de la varicela como infección primaria y del herpes zoster como enfermedad recurrente. Puede afectar el SNC tanto durante la infección primaria como durante la recurrencia. Como todos los herpesvirus, luego de la infección primaria la persona permanece infectada de manera latente para toda la vida. El VVZ establece la latencia en los ganglios de la raíz dorsal, entéricos y autónomos a lo largo del neuroeje;41 pero al ocurrir su reactivación replicativa causa diversas complicaciones neurológicas (cuadro). La cerebelitis aguda es una de las enfermedades cerebelosas más frecuentes asociadas con el VVZ en la infancia y raras veces es informada en adultos, en que puede estar acompañada o no de erupciones cutáneas. La fisiopatología de la encefalitis o cerebelitis por VVZ difiere entre pacientes con erupciones y sin estas. En pacientes con erupción cutánea, producto a la replicación previa del virus y el aumento de la carga viral, la infección causa encefalitis en un período corto, debido a la propagación del patógeno al SNC a través del transporte axonal o por medio del torrente sanguíneo. En pacientes sin erupción cutánea, el virus se reactiva dentro de los ganglios sensoriales espinales y se propaga al SNC mediante transporte axonal, lo que facilita la invasión e infección del encéfalo.42 Además de la invasión e infección directa del virus, mecanismos autoinmunes también están involucrados en las neuropatologías. Por ejemplo, se informa la presencia de anticuerpos antirreceptor NMDA, lo cual afecta la neuroplasticidad y la transmisión sináptica.43 También se informa en niños con ataxia aguda posinfecciosa por VVZ la presencia de anticuerpos anticentrosomas que reaccionan contra la pericentrina. Las células de Purkinje contienen numerosas pericentrinas y más de un centrosoma, por lo que son muy vulnerables al ataque de estos anticuerpos y por tanto, están involucrados en la degeneración cerebelosa y el desarrollo de la ataxia posinfecciosa.44
Este virus es el más asociado a la ataxia cerebelosa aguda y la ataxia posinfecciosa aguda en niños. Estudios realizados revelaron desmielinización y cambios inflamatorios en el cerebelo, así como una reducción anormal en el flujo sanguíneo regional del cerebelo. Estas afectaciones pueden estar implicadas en las manifestaciones atáxicas.45 Sin embargo, hay pocos informes de cerebelitis por VVZ en adultos, confirmado por exámenes virales del LCR.42 Estudios en pacientes adultos informan lesiones en el tallo cerebral y el puente46 e hiperperfusión del cerebelo.42 En individuos con edad avanzada e inmunodeficientes, la inmunidad mediada por células contra el VVZ disminuye, lo que permite la reactivación viral del VVZ y causa daños cerebelosos.47
Virus Epstein-Barr (VEB)
El VEB puede infectar directamente el SNC, lo cual se evidencia por la detección de anticuerpos contra VEB en el LCR.7 Además el antígeno de la cápsida (EBNA) comparte un pentapéptido con la proteína básica de la mielina, por lo que la respuesta inmunitaria de linfocitos T CD4+ contra el virus produce desmielinización dado por esta semejanza estructural.48 También los linfocitos T se activan al reconocer en la superficie de los linfocitos B infectados por VEB la proteína alfa-B cristalina, que es una proteína de estrés térmico responsable de la respuesta inmune por linfocitos T. En circunstancias en que los linfocitos T activados acceden al SNC se produce un ataque contra las células gliales que expresan alfa-B cristalina.49 En otro estudio se señala que VEB daña los ganglios basales directamente, lo cual provoca la calcificación de estos,50 mientras que la infección por VEB puede facilitar la infiltración cerebral por la activación de macrófagos y linfocitos.51
Aunque las complicaciones neurológicas por infección por VEB son raras (cuadro), tienen una incidencia del 0,37 % al 7,3 % de los pacientes infectados.52 Estas afectaciones causan daños en el SNC tales como la hidrocefalia, hipoperfusión en el cerebelo y cerebelitis.53 Se describen procesos inmunopatogénicos relacionados con la ataxia cerebelosa posinfecciosa aguda causada por VEB. En pacientes con ataxia posinfecciosa se han detectado anticuerpos IgG e IgM contra elementos nucleares y citoplásmicos de las neuronas.54 También se informan altos títulos de anticuerpos contra los receptores de glutamato δ2 en el LCR,55 receptores que se expresan predominantemente en las células de Purkinje y que desempeñan un papel clave en la plasticidad sináptica y la coordinación motora. No obstante, se desconoce el mecanismo exacto de la ataxia cerebelosa aguda mediada por VEB, puede ser resultado de la invasión directa del virus al cerebelo, de un fenómeno autoinmune posinfeccioso; o incluso de la combinación de ambos. La detección de anticuerpos contra VEB en el LCR apoya la infección viral directa del cerebelo.52 Por el contrario, se plantea que es más probable un mimetismo molecular debido a la presencia de estos autoanticuerpos, en lugar del efecto viral directo, pues en casos similares no se ha encontrado VEB en el LCR lo que pudiera reflejar la etiología no infecciosa de la enfermedad.9
Virus dengue
La patogenia de las complicaciones neurológicas en la infección por el virus dengue no está esclarecida. La invasión directa del virus y los mecanismos autoinmunes son causantes del daño neuronal, aunque aún no se describe un mecanismo neuropatogénico exacto.56 A pesar de que el virus dengue inicialmente se consideró no neurotrópico, se comprobó que infecta neuronas de ratón cultivadas in vitro. El receptor de membrana parece facilitar la invasión solo para neuronas específicas, pues los astrocitos no resultaron infectados en ese experimento.57 Posteriormente, la neuroinvasión se demostró mediante la detección de antígenos del virus en el cerebro por inmunohistoquímica en casos fatales de encefalopatía, y también por la detección de anticuerpos IgM antidengue en el LCR en pacientes con encefalitis;58 también se ha encontrado ARN viral en el LCR y en el tejido cerebral de casos fatales.59,60
Las complicaciones neurológicas en la infección por el virus dengue pueden ser explicadas por tres mecanismos patógenicos: la invasión directa del virus al SNC; complicaciones sistémicas debido a complicaciones metabólicas como el desequilibrio electrolítico, liberación de productos tóxicos e insuficiencia hepática y renal; y efectos inmunológicos provocados después de la infección.58,61 En modelos animales la infección produce daños severos al SNC como hemorragia, gliosis reactiva, microglia hiperplásica e hipertrofiada, proliferación de astrocitos y retracción de las neuronas de Purkinje, resultado de la replicación viral en las neuronas, astrocitos, microglia y oligodendrocitos.60
Varias manifestaciones neurológicas (cuadro) se informan en la infección por el virus dengue; las más comunes son la encefalitis y la encefalopatía. Cualquier serotipo del virus puede provocar estas complicaciones neurológicas, pero los serotipos 2 y 3 son los involucrados con mayor frecuencia en las enfermedades neurológicas graves.58 Los casos de trastornos del movimiento son raros, se informa parkinsonismo,59 SOMA;6 ataxia por encefalomielitis aguda diseminada posinfecciosa;62 ataxia por vasculitis cerebral61 o por cerebelitis.56 El SOMA puede ser resultado de un mecanismo autoinmune posinfeccioso, sin embargo, la invasión viral directa del cerebro y cerebelo no puede ser excluida, debido a las evidencias que revelan el neurotropismo del virus.6 En la encefalomielitis aguda diseminada, en que se informan casos con ataxias,62 la fisiopatología implica una respuesta autoinmune transitoria dirigida a la mielina u otros antígenos propios, posiblemente por una activación inespecífica de clones de células T auto reactivos o por mimetismo molecular.63 En este caso se informan lesiones de materia blanca en el centro semioval, la corona radiata, el tálamo; desmielinización en la médula espinal; entre otros.58,62 En la ataxia por cerebelitis se informan daños en la corona radiata, en el lóbulo frontal izquierdo y en el cerebelo.56 Un estudio informa un caso de ataxia causado por el virus dengue, producto de una vasculitis cerebral, en el cual se encontraron infartos múltiples que afectaban a las regiones del puente y la médula.61
Virus del Nilo Occidental (VNO)
Se estima que aproximadamente el 1 % de las infecciones con el VNO resulta en una enfermedad neuroinvasiva grave.64 El VNO causa parálisis flácida aguda, así como la afectación focal del parénquima del cerebelo, el tallo encefálico, el rombencéfalo y la médula espinal. También se le atribuye trastornos del movimiento (cuadro).65
Los mecanismos neuropatogénicos para VNO no están aclarados; se proponen vías hematógenas y transneurales.66 La citocina proinflamatoria osteopontina, compromete la integridad de la BHE mediante el reclutamiento y la infiltración de neutrófilos polimorfonucleares infectados con VNO, y facilita la entrada del virus al SNC a través del mecanismo caballo de Troya.67 El daño al SNC es resultado además de la respuesta inmune a la infección por el virus en las células residentes en el SNC y/o por los leucocitos infiltrados después de las respuestas inmunes sistémicas. Además, durante la infección con VNO, se incrementa la expresión de la senataxina helicasa humana con una respuesta antiviral notable, pero esta enzima está relacionada con ataxias con apraxia oculomotora tipo 2.68,69
El VNO rara vez es la causa de la ataxia cerebelosa aguda; varios informes notifican casos del SOMA causado por el VNO. Dado que la mayoría de los pacientes informados no mostraron lesiones supratentoriales en la RMN, el estado mental alterado y los cambios en el electroencefalograma podrían interpretarse en el contexto de una disfunción de la sustancia gris inducida por la infección, o en el contexto de un deterioro de la red cerebelo-cortical. Investigaciones demuestran que las lesiones focales del tallo cerebral pueden causar hipoperfusión cortical generalizada con disfunciones secundarias.65 Un estudio que comparó la volumetría cerebral de controles sanos y pacientes con opsoclonia mostró una disminución del volumen de la corteza cerebelosa con la preservación del volumen de la sustancia blanca del cerebelo y una disminución posterior de la materia gris cortical y subcortical de los hemisferios cerebrales.70
Enterovirus. Enterovirus 71 (EV71)
Los enterovirus son conocidos por causar la enfermedad de manos, pies y boca, y la poliomielitis. Su principal vía de transmisión es fecal-oral. Algunos enterovirus tienen la capacidad de infectar el SNC y causar síntomas neurológicos. El poliovirus es el patógeno más conocido por causar poliomielitis en niños, provocada por la replicación viral en las neuronas motoras de la médula espinal. Aunque este virus está en vía de erradicación, hay algunos países que aún tienen brotes, principalmente derivados de vacunas basadas en el virus. Otros enterovirus no polio causan aproximadamente la mitad de los casos de meningitis aséptica en niños. La infección por los virus coxsackie y echovirus es la causa más común de meningitis viral, aunque también el poliovirus y el EV71 provocan meningitis aséptica. EV71 es uno de los enterovirus más comunes que se han aislado de las muestras recogidas de niños con encefalitis viral y es causante de brotes de meningitis graves y meningoencefalitis.71
EV71 afecta al SNC infectándolo directamente, pues las partículas virales pueden ser aisladas del LCR.72 Dado que EV71 puede infectar varias células inmunes, como las células CD14+, las células dendríticas y las células mononucleares de sangre periférica, es posible que EV71 use estas células como vehículos para invadir al SNC.71 Como un mecanismo neuropatogénico, se informa la interacción del virus con el receptor de captación B2 (SCARB2) presente en la mielina, lo que induce desgranulación y daño neuronal en el tallo cerebral.73 Las complicaciones neurológicas causadas por EV71 se muestran en el cuadro.74 La encefalitis del tallo cerebral es la manifestación neurológica más crítica, pues puede causar hemorragia o edema pulmonar neurogénico que conduce a la muerte.73
Las afectaciones neurológicas más prominentes del SNC causadas por EV71 son la sacudida mioclónica (68 %), ataxia (35 %) y temblor (21 %).75 La ataxia aparece más frecuente asociada a la encefalitis del tallo cerebral,76 en que existen lesiones del núcleo dentado del cerebelo y del tallo cerebral reveladas en la RMN.77 El SOMA se considera una manifestación rara de EV71. La causa más probable se debe a mecanismos inmunes posinfecciosos, en lugar de una infección directa, pues hay estudios en que no se detecta EV71 en el LCR;78 aunque la infección directa no puede ser descartada, debido a que este síndrome está asociado con daños al tallo encefálico y el núcleo dentado del cerebelo, donde este virus puede invadir.
Diagnóstico de las ataxias infecciosas
El diagnóstico de las ataxias infecciosas se basa primeramente en el examen neurológico y neuroimagen. Dificultades en la marcha, alteración del equilibrio, nistagmo, dismetría y dificultad para hablar son manifestaciones clínicas características.79 Los exámenes virológicos juegan un papel importante en este diagnóstico, pues permiten confirmar si la afectación neurológica se debe realmente a una etiología viral. Los métodos para confirmar la infección por estos virus implican la detección de partículas virales, del ácido nucleico viral y antígenos o anticuerpos, ya sea en el suero o en el LCR de los pacientes. El aislamiento viral, la detección del ácido nucleico viral o de antígenos virales se emplean en la confirmación de la infección en fases tempranas de la enfermedad; en fases tardías, la serología es más adecuada para el diagnóstico, pues la sensibilidad del aislamiento y la reactividad antigénica disminuyen.58
El aislamiento viral es la prueba de oro de la confirmación de la infección. Sin embargo, la detección del ácido nucleico viral mediante el ensayo de reacción en cadena de la polimerasa (PCR) suele ser más empleada, pues es una técnica muy sensible, específica y consume menos tiempo. Con este método es posible detectar el genoma viral o las partículas virales en LCR, suero y biopsias del tejido cerebral.46,65 Estos dos métodos son muy costosos y laboriosos, pero también son más específicos que los métodos serológicos utilizados para la detección de anticuerpos. La detección de los virus en biopsias del tejido cerebral es otro método que permite identificar el agente etiológico causante de las ataxias infecciosas.74 La detección de anticuerpos IgM específicos para los virus mediante ensayos inmunoenzimáticos es el método serológico de elección para el diagnóstico en la fase temprana de la enfermedad.36,58 La seroconversión de IgM y el aumento del título de IgG en sueros pareados confirman la infección aguda. Estos anticuerpos no solo se encuentran en sangre, sino también en el LCR de los pacientes. El ensayo de neutralización viral in vitro es otro método utilizado para el diagnóstico de estos virus.
Conclusiones
Las ataxias infecciosas causadas por virus neurotrópicos siguen siendo enfermedades raras y poco estudiadas. Aunque la mayoría de los informes de casos abordados refieren la evolución satisfactoria de los pacientes, hay infecciones que dejan secuelas neurológicas e incluso conducen a la muerte. Los mecanismos neuropatogénicos exactos que se relacionan con esta ataxia aún no están esclarecidos en la actualidad, aunque se sugieren procesos neuroinflamatorios como la principal causa. Lo anterior indica la necesidad de realizar estudios futuros que profundicen la caracterización clínica, neurofisiológica, neuropatológica y molecular de esta afectación para favorecer su diagnóstico y tratamiento.

