










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La leucemia mieloide aguda (LMA) es un trastorno genéticamente heterogéneo, caracterizado por la adquisición somática de alteraciones genéticas y epigenéticas en células madres progenitoras hematopoyéticas que alteran sus mecanismos normales de auto-renovación, proliferación y diferenciación celular. En dependencia de las características del clon celular que prolifera se observará diferentes comportamientos clínico, hematológico y citomorfológico.1,2,3
Esta gran variabilidad impulsó la necesidad de establecer subgrupos según las características que se manifiestan. El primer sistema integral para clasificar a la LMA se estableció en 1976 y fue elaborado por el Grupo Cooperativo Franco-Américo-Británico (FAB).4 Esta clasificación buscaba identificar el equivalente leucémico de las células mieloides para cada estadio normal de diferenciación atendiendo a criterios morfológicos y citoquímicos. Sin embargo, a pesar de su utilidad diagnóstica y constituir la primera herramienta a la mano de los investigadores, se reconoció que la interpretación morfológica era insuficiente para establecer los grupos de riesgo de manera adecuada. El conocimiento cada vez más profundo de la biología de la LMA, puso de manifiesto la necesidad de incorporar nuevos parámetros para una correcta agrupación. De esta manera, en el año 2002 surge la clasificación de la Organización Mundial de la Salud (OMS),5 que incorpora e interrelaciona las características morfológicas, citogenéticas y moleculares, y busca ser no sólo una herramienta útil para el diagnóstico sino también para la clínica al correlacionarse de una forma más precisa con el pronóstico. En los años 2008 y 2016, la OMS ha actualizado la clasificación,6,7 incorporando cada vez nuevas anomalías citogenéticas y moleculares definitorias del diagnóstico diferencial de las LMA.
En este sistema de clasificación, los pacientes con translocaciones como la t(8;21)(q22;q22) y t(15;17)(q22;q21), así como con la inversión del cromosoma 16 (inv(16)(p13q22)( o translocación t(16;16)(p13;q22), fueron clasificados como “LMA con anomalías citogenéticas recidivantes”. Estas aberraciones cromosómicas corresponden a las anomalías moleculares conocidas como genes de fusión RUNX1-RUNX1T1 (nombrado antes AML1-ETO), PML-RAR( y CBF(-MYH11 respectivamente. Determinar su presencia a través del estudio citogenético o molecular permite enmarcar al paciente en un subtipo específico de LMA.
Las LMA portadoras de las aberraciones cromosómicas t(8;21)(q22;q22) e inv(16)(p13q22)/t(16;16)(p13;q22), con las que se forman los reordenamientos de genes antes mencionados, son definidas como LMA CBF (CBF, sigla del inglés de factor de unión al núcleo). Esta entidad está entre los subtipos más comunes de LMA, ya que las dos aberraciones juntas aparecen aproximadamente en el 25 % de los pacientes pediátricos8 y entre el 15 % a 20 % de los adultos, con LMA de novo.8,9 Sin embargo, aunque las LMA CBF están agrupadas bajo un mismo nombre, molecularmente son bien diferentes, lo cual conduce a comportamientos clínicos particulares.10
La biología molecular también ha permitido encontrar anomalías que no implican rearreglos cromosómicos. Estas son mutaciones que modifican la secuencia del ADN y que han cobrado importancia para la caracterización y la predicción de riesgo de las LMA, sobre todo en aquellas con cariotipo normal (CN). Ejemplo de ello son: la duplicación interna en tándem del gen FLT3 (DIT FLT3), las mutaciones en los genes FLT3, NPM1, CEBPα y C-KIT, y las mutaciones en marcadores epigenéticos.11
El FLT3 es un receptor de membrana, con un dominio tirosina quinasa intracelular, que normalmente está expresado en células madres progenitoras hematopoyéticas y juega un importante papel en los estadios tempranos del desarrollo de las líneas linfoide y mieloide.12) La DIT FLT3 es la mutación más frecuente del gen que codifica al mencionado receptor y se manifiesta en una longitud variable, desde tres hasta más de 400 pares de base. La aberración está localizada en el dominio yuxta membrana y conduce a la activación constitutiva del receptor; lo cual promueve la proliferación celular y el bloqueo de la diferenciación de progenitores hematopoyéticos tempranos.13) En general, se plantea que aparece en alrededor del 25 % de todos los casos de LMA y que su frecuencia varía muy poco con la edad.12 La DIT FLT3 se relaciona con un resultado adverso en las poblaciones adultas y pediátricas con LMA. Su presencia predice la ocurrencia de una alta tasa de recaída, lo que se traduce en una sobrevida media inferior respecto al resto de pacientes con LMA.14,15 Por todo lo anterior, su estudio ha adquirido un papel crítico en la estratificación de riesgo.
Las mutaciones del gen NPM1 aparecen con alta frecuencia en los pacientes con LMA, 25 a 53 %, y más aún en las LMA con CN en que pueden estar entre un 46 a 67%.16 Se han descrito más de 50 variantes, siendo la NPM1-A la que más se encuentra en pacientes adultos; mientras que en pacientes pediátricos se informa mayoritariamente la variante “B”.17 Más del 95 % de las mutaciones consisten en una inserción de cuatro pares de base en la posición 863 que generan modificaciones en el extremo C terminal de la proteína y provocan su localización aberrante en el citoplasma.14 Estudios in vivo en modelos de ratones han demostrado que NPM1 es un gen supresor de tumor haploinsuficiente y que por tanto, la pérdida de su función puede contribuir a la patogénesis de la LMA.18 A pesar de que las mutaciones en NPM1 se asocian a una evolución favorable, su sola presencia no determina el pronóstico ya que su asociación con otras mutaciones en genes como FLT3, DNMT3A, NRAS, IDH y PTPN11 determinan su estratificación.16
En los últimos años se han identificado alteraciones en los genes implicados en la regulación epigenética, que se consideran una tercera clase de mutaciones y muestran efectos en la diferenciación celular y la proliferación. Estas mutaciones, que pueden ser identificadas en el 40 % de los casos de LMA, actúan sobre genes que tienen relación con la metilación del ADN como el DNMT3A, el TET2, el IDH-1 y el IDH-2.1
Para la determinación de muchas de las anomalías es necesaria la secuenciación, lo cual limita generalizar su estudio debido a su alto costo. Sin embargo, la reacción en cadena de la polimerasa (PCR por su sigla en inglés) como técnica sensible, específica, rápida, sencilla y de relativamente bajo costo, ha permitido introducir el estudio molecular como herramienta de rutina para el diagnóstico clínico. Debido a la frecuencia con que aparecen y la importancia que tienen para el pronóstico, se han diseñado protocolos de PCR para la detección de algunas aberraciones moleculares que inicialmente requerían ser secuenciadas, entre ellas la DIT FLT319 y la NPM1-A (mutación “A” del gen NPM1).20
Desde el año 1985 en el Instituto de Hematología e Inmunología “Dr. José Manuel Ballester Santovenia” (IHI) se ha introducido progresivamente el estudio molecular de las hemopatías malignas.21 Actualmente, los pacientes con LMA se benefician de la determinación por PCR de cinco de las anomalías más importantes para la definición diagnóstica y pronóstica de esta entidad. Anteriormente, se ha reportado la incidencia del gen de fusión PML-RAR( en la LPM22 y del RUNX1-RUNX1T1,23-24 en pacientes cubanos con LMA. En el más reciente de los citados informes, se correlacionó la presencia simultánea del RUNX1-RUNX1T1 con otras alteraciones moleculares (CBF(-MYH11, DIT FLT3 y NPM1-A). Sin embargo, no ha sido informada la frecuencia con que aparecen los genes de fusión RUNX1-RUNX1T1 y CBF(-MYH11, la DIT FLT3 y la mutación NPM1-A en pacientes con LMA.
El objetivo de esta investigación fue determinar la frecuencia de estos cuatro biomarcadores, en pacientes cubanos con leucemias mieloides agudas primaria no promielocíticas.
Métodos
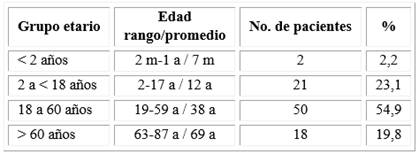
Se estudiaron 91 pacientes entre julio de 2013 a julio de 2016. Se tuvieron en consideración los protocolos de tratamiento establecidos según la edad al debut, los pacientes fueron clasificados en cuatro grupos etarios: menores de dos años, de dos a menos de 18 años, de 18 a 60 años y mayores de 60.
A partir de sangre medular se aisló ARN y se obtuvo ADN complementario mediante transcripción inversa. Se amplificaron los fragmentos correspondientes a cada alteración molecular mediante PCR a partir de tres protocolos diferentes: uno para los genes de fusión RUNX1-RUNX1T1 y CBF(-MYH11,25 otro para la DIT FLT319 y un tercero para la mutación NPM1-A.20 Los programas de amplificación fueron los recomendados en cada protocolo y el análisis del producto de la PCR se realizó mediante electroforesis capilar en todos los casos. Para el procesamiento y análisis de los datos se empleó el programa Epidat 3.1. Las cuatro alteraciones moleculares no fueron estudiadas en todos los pacientes, por lo que el análisis se realizó de acuerdo al número de pacientes en que fue estudiada cada una de ellas.
Resultados
En la tabla 1 se muestra la frecuencia total con que se encontraron las cuatro alteraciones moleculares respecto al total de pacientes estudiados en cada una de ellas.
Tabla 1 Frecuencia general de las alteraciones moleculares

% de pacientes positivos respecto a la “n” para cada alteración
La distribución por edades y el promedio de edad por grupo se muestra en la tabla 2. La edad promedio entre los pacientes fue de 30 años y el rango de edad estuvo entre dos meses y 87 años.
La figura 1 muestra la distribución por edad en las alteraciones moleculares. No se muestra el grupo de niños menores de dos años (n= 2). Los porcentajes fueron calculados de acuerdo al total de pacientes en que cada alteración molecular pudo ser estudiada.
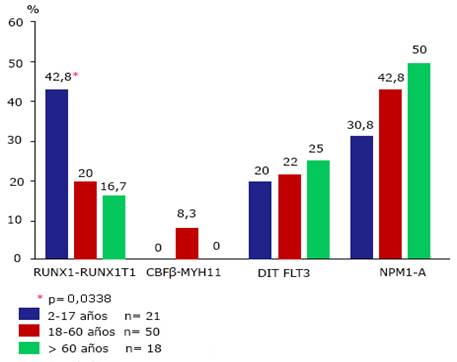
Fig Frecuencia de las alteraciones moleculares RUNX1-RUNX1T1, CBF(-MYH11, DIT FLT3 y NPM1-A en pacientes cubanos con leucemia mieloide aguda primaria no promielocítica clasificados en tres grupos etarios.
El análisis estadístico reveló que solo la presencia del RUNX1-RUNX1T1 tuvo relación con la edad (Fig.). Su frecuencia disminuyó en la medida que la edad aumentó y la diferencia fue significativa (p( 0,0338) entre el grupo de niños y los adultos. La diferencia entre ambos grupos de adultos, aunque mostró tendencia a disminuir hacia edades mayores, no fue significativa. El gen de fusión CBFβ-MYH11 solo se encontró entre los adultos menores de 60 años. Mientras que la DIT FLT3 tuvo una presencia casi uniforme en los tres grupos analizados. En el caso de la NPM1-A, aunque se observó una tendencia a aumentar con la edad, las diferencias no fueron significativas (p> 0,05). Su presencia fue mayoritaria entre los pacientes adultos. Mientras que en el grupo de niños la aberración más frecuente fue el gen de fusión RUNX1-RUNX1T1.
En algunos pacientes se observó la existencia simultánea de alteraciones. En 15 pacientes hubo más de una alteración. La NPM1-A y la DIT FLT3 aparecieron simultáneamente en nueve pacientes, resultando ser la combinación más frecuente. En algunos de los pacientes portadores del RUNX1-RUNX1T1 aparecieron también la NPM1-A (n( 3) o la DIT FLT3 (n( 2). Un paciente fue positivo a estas tres alteraciones. Mientras que el gen de fusión CBFβ-MYH11 se observó siempre como alteración única.
Discusión
El estudio molecular de las LMA permite ajustar el diagnóstico a las recomendaciones de la clasificación más actual para de esta manera aplicar medidas terapéuticas de acuerdo a cada paciente. También permite la estratificación según el riesgo y, a largo plazo, realizar una correlación con la respuesta al tratamiento para evaluar el pronóstico y la supervivencia.
Se conoce que la LMA se manifiesta mayormente en adultos y entre ellos es más frecuente en los mayores de 60 años.26,27,28 Sin embargo, en el presente estudio la mayor cantidad de adultos tuvo menos de 60 años. Es posible que muchos de los pacientes de avanzada edad, debido al alto riesgo que para ellos representa la LMA,27 no alcancen a ser estudiados. Como era esperado, la cantidad total de pacientes pediátricos fue menor al total de adultos; tal fue así que la baja incidencia de niños menores de dos años no permitió realizar su análisis. Sólo pudo ser analizado el grupo de niños de dos años en adelante. Debe señalarse que esta investigación incluyó sólo a los pacientes con estudio molecular por lo que la muestra no representa la incidencia de la LMA en Cuba.
Desde el año 2002 la OMS incluyó, entre otros, dos subtipos específicos de LMA caracterizados por los genes de fusión RUNX1-RUNX1T1 y CBF(-MYH11. Como se conoce, estos se forman a partir de aberraciones cromosómicas estructurales y su presencia identifica un grupo de LMA denominado: LMA CBF. Aunque las LMA CBF se asocian a una evolución favorable; su coexistencia con otras anomalías citogenéticas y moleculares, así como otros elementos clínicos específicos influyen de manera diferente en la evolución de estos pacientes.29,30 De aquí que para predecir el riesgo en las LMA CBF, es importante realizar un análisis integral clínico, genético y molecular. Se ha encontrado que las fusiones que afectan el CBF pueden aparecer a cualquier edad pero tienen mayor incidencia en niños y adultos jóvenes.31
RUNX1-RUNX1T1
En este informe el gen de fusión RUNX1-RUNX1T1 fue más frecuente en la edad pediátrica y así lo describen también otros autores.30,32,33 La diferencia, que fue significativa entre niños y adultos, se corresponde con lo reportado por Bolouri y otros,34 respecto a que aparece un pico de frecuencia en la edad pediátrica que disminuye con la edad.
En general, se plantea que el gen de fusión RUNX1-RUNX1T1 está presente aproximadamente entre el 5 % y 12 % de todos los casos de LMA.32,35,36 El porcentaje superior encontrado, tanto para el total de pacientes, como cuando fueron estratificados de acuerdo a la edad, pudiera estar relacionado con la no inclusión de pacientes con el subtipo LPM. Similar al presente es el caso del estudio de Kihara y otros,37 que notifican una frecuencia del 20,8 % en un estudio en adultos donde no incluyen a la LPM, como en la presente investigación.
CBFβ-MYH11
La escasa presencia del gen de fusión CBFβ-MYH11 en este trabajo estuvo en correspondencia con lo descrito por otros autores.38,39,40,41 El CBFβ-MYH11 apareció con una frecuencia similar a la encontrada por Slovak y otros,39 pero otros autores refieren frecuencias menores.40,41) En este sentido, Vaskova y otros38 no encontró alteración en 90 pacientes adultos. Otros autores informaron frecuencias muy parecidas entre el RUNX1-RUNX1T1 y el CBFβ-MYH11. Sin embargo, en el presente estudio se encontró una frecuencia mucho menor del CBFβ-MYH11 respecto al RUNX1-RUNX1T1, lo que marca una diferencia con lo planteado en la literatura.
En los pacientes pediátricos de este trabajo, el comportamiento de la fusión CBF(-MYH11 difiere de otros autores que sí lo encuentran.
NPM1
La presencia de mutaciones en el gen NPM1 osciló en un rango de entre 25 % a 53 % en todas las LMA.18 Los artículos coinciden en que estas son las que con mayor frecuencia aparecen en esta entidad. Ejemplo de esto es el estudio realizado recientemente por Yusoff y otros,14 los cuales estudiaron las mutaciones en los genes FLT3 y NPM1 de pacientes con LMA. Mediante la secuenciación del gen NPM1 encontraron que el 27,1 % de los pacientes presentaba alguna mutación; el porcentaje que resultó mayor al encontrado para las mutaciones en el gen FLT3 (DIT-FLT3 y mutaciones en el dominio tirosina quinasa de FLT3). En el presente trabajo, en que solo se estudió la NPM1-A, esta resultó ser la aberración más frecuente en el total de pacientes. Al analizar por grupos etarios, esta fue igualmente mayoritaria en los pacientes adultos; este hallazgo coincide con la literatura que identifica a la variante “A” como la que más se encuentra en pacientes adultos.17 Sin embargo, de manera similar a otros informes, no ocurrió lo mismo en los pacientes pediátricos; para estos se plantea que la mutación “B” y no la “A” es la que más se manifiesta.17
DIT FLT3
La DIT FLT3 puede aparecer en todos los subtipos de LMA. Se reportan frecuencias de aparición diferentes, desde un 20 hasta 37 % en el adulto.14,42,43,44,45,46,47,48,49 Mientras que en niños algunos autores encuentran frecuencias algo menores (entre el 10 y el 20 %, aproximadamente),50,51,52 otros plantean que su frecuencia varía muy poco con la edad12 y notifican que la DIT FLT3 aparece en alrededor del 25 % de todos los casos de LMA.12,53
La presencia casi uniforme de la DIT FLT3 que se observó, así como la magnitud de las frecuencias en los tres grupos etarios está en correspondencia con la mayoría de autores, sobre todo con aquellos que encuentran que su frecuencia varía muy poco con la edad.
Coexistencia de alteraciones moleculares
La coexistencia de alteraciones moleculares apoya la hipótesis de “doble impacto” (conocida en inglés como “two-hits”) acerca de la leucemogénesis en la LMA.54) Esta plantea que el desarrollo de la entidad requiere la presencia de más de una alteración molecular, casi siempre de una mutación de Clase I que aportan ventajas proliferativas o de supervivencia como la DIT FLT3, con otra de Clase II (RUNX1-RUNX1T1, CBFβ-MYH11 y NPM1, entre otras) que alteran la diferenciación celular y la apoptosis. No obstante, también pueden coexistir las de Clase II entre sí.
Duployez y otros,8 en un estudio de mutaciones en las LMA CBF encontraron concomitancia en más del 90 % de los pacientes. El fenómeno lo observaron tanto en presencia del RUNX1-RUNX1T1 como del CBFβ-MYH11 y sus pacientes presentaron desde dos hasta seis aberraciones juntas. En la presente investigación no se dispuso del estudio de las cuatro alteraciones en todos los pacientes por lo que no es válido analizar cuantitativamente el comportamiento de la concomitancia. Sin embargo, vale destacar la ocurrencia del fenómeno, pues apoya la hipótesis multifactorial de la leucemogénesis de la LMA. La concomitancia de la mutación NPM1-A y la DIT FLT3, de Clase II y I respectivamente, fue la más frecuente. Precisamente acerca de esta combinación Liu y otros 55 plantean que las mutaciones en NPM1, además de estar asociadas frecuentemente a CN, también lo están con frecuencia a la DIT FLT3. Respecto a esto, Falini y otros,56 describieron en 2005 que alrededor del 40 % de las LMA con NPM1 mutado también presentan la DIT FLT3. Como se mencionó, la concomitancia de tres o más alteraciones moleculares está descrita;57 en este estudio se presentaron tres en un paciente, el RUNX1-RUNX1T1, la NPM1-A (ambas de Clase II) y la DIT FLT3. Aunque no se encontró coexistencia del gen de fusión CBFβ-MYH11 (mutación de Clase II) con otra alteración otros autores sí la reportan; ejemplo de ello es su asociación con mutaciones en los genes KRAS, NRAS y KIT encontrada por Paschka y otros.58
Con anterioridad, ha sido descrita la coexistencia del gen de fusión RUNX1-RUNX1T1 con las aberraciones CBFβ-MYH11, DIT FLT3 y NPM1-A en un grupo de pacientes cubanos con LMA.24 Sin embargo, el presente análisis no se centró solo en los pacientes positivos al RUNX1-RUNX1T1; por tanto, este representa el primer reporte de frecuencia de las cuatro aberraciones en pacientes cubanos con LMA no LPM.
Por primera vez se describe la frecuencia de los cuatro biomarcadores moleculares en pacientes cubanos con leucemias mieloides agudas primaria no promielocíticas; su comportamiento fue similar a lo descrito por otros autores, aunque se encontraron algunas particularidades.

