










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC vol.9 no.2 Camagüey mar.-abr. 2005
CASOS CLÍNICOS
Fibrosis quística. Informe de un caso
Cystic fibrosis. Case report
Dra. Olga Lidia García Peña; Dra. Hortensia Aguirre del Busto; Dra. Esther Llano Padrón; Dra. Norma Hernández Gómez; Dra. Nancy Ratón Quintana
Hospital Provincial Pediátrico Docente Eduardo Agramonte Piña. Camagüey, Cuba.
RESUMEN
La fibrosis quística es una enfermedad genética hereditaria crónica, multisistémica y potencialmente letal que se caracteriza por afección pulmonar con infecciones sobreañadidas y recurrentes, insuficiencia pancreática que origina trastornos nutricionales y un fallo en el crecimiento.
Se presenta un caso portador de fibrosis quística, homocigótico para la mutación mayor (pruebas genéticas moleculares) Df508 / Df508, el cual no mostró las manifestaciones comunes de esta afección, ya que presentó a los 49 días de nacido una anemia severa refractaria a tratamiento, lo que motivó su hospitalización, estudio y tratamiento hasta sus conclusiones.
DeCS: FIBROSIS QUÍSTICA; ANEMIA; LACTANTE; INFORME DE CASO.
ABSTRACT
Cystic fibrosis is a genetic chronic hereditary multisystemic and potencially lethal disease which is charaterized by pulmonary affection with recurrent infections, pancreatic failure originating nutritional disorders and a growing deficiency. It is presented a case, carrier of cystic fibrosis, homocygotic for the major mutation (molecular genetic tests) Df508/Df508 who did not show common manifestations of this disease because he presented at days of birth; this motivated his hospitalization, study and treatment until his conclusions.
DeCS: CYSTIC FIRBOSIS; ANEMIA; INFANT; CASE REPORT.
INTRODUCCIÓN
La fibrosis quística (FQ) es una vieja enfermedad genética crónica incurable, hereditaria, multisistémica y potencialmente letal que se caracteriza por presentar una afección obstructiva pulmonar con infecciones sobreañadidas y recurrentes, insuficiencia pancreática que origina trastornos nutricionales con fallo en el crecimiento y cifras de los electrolitos en el sudor elevados. 1
El tipo de herencia es autosómico recesiva, no ligada al sexo con una incidencia variable, 1 por cada 2500 nacidos vivos en países de raza blanca, 1 por cada 4500 y hasta 5000 en países con mestizaje. 1
El gen de la FQ fue localizado en el brazo largo del cromosoma 7, en el año 1985 y aislado en 1989, esta identificación del gen marcó un hito en la historia de esta enfermedad, se describe una mutación mayor Df508 2
Desde el descubrimiento del gen, más de 700 mutaciones han sido descritas, pero no todas pueden ser demostradas, en Cuba se estudian cuatro de ellas. 2
Por eso la sospecha y evaluación clínica, unido al estudio del electrolito en el sudor, son los elementos más importantes para el diagnóstico, además por la fisiopatología y la utilización del tratamiento multidisciplinario no cabe duda que la supervivencia de estos pacientes se ha incrementado, unido a ello el nivel de conocimiento de nuestros médicos y la voluntad política que existe en nuestro país hacia la salud pública en general y en particular hacia la FQ y otras enfermedades hereditarias, la cual ha permitido que estos niños dispongan hoy del seguimiento y tratamiento adecuado multidisciplinario que esta afección necesita.
Se presenta un caso portador de FQ homocigótico para la mutación mayor, el cual no mostró las características clínicas antes señaladas, manifestó una anemia severa 79g/l que ocasionó su hospitalización y estudio.
PRESENTACIÓN DEL CASO
Paciente PRC, de 49 d de nacido, con HC 494464 que fue remitido de su área de salud con Hb 83g/l, cuando se rectificó en el hospital presentaba 79 g/l sin repercusión hemodinámica.
Antecedentes de embarazo normal, parto eutócico a las 36 semanas con peso referido de 6, 4 libras, aproximadamente 2950 g, a la semana del nacimiento presentó íctero fisiológico, el cual no manifestó en el momento del ingreso.
Fecha de nacimiento: 2 de septiembre de 2001.
Primer ingreso: 19 de octubre de 2001.Peso: 3450 g. Lactante con buen estado general, palidez cutáneo mucosa intensa, hidratado, no edemas, no dificultad respiratoria, murmullo vesicular normal, no estertores, frecuencia respiratoria 56 x min., tonos cardíacos rítmicos bien golpeados, no soplos, no cianosis, buenos pulsos, no gradiente térmico, frecuencia cardiaca 152 x min, no presentó visceromegalias a la palpación, no íctero, lactancia materna exclusiva. No se realizaron otros estudios en este ingreso.
Se interpretó como anemia carencial, se transfundió y se dio de alta con Hb 87g/l.
Segundo ingreso: 30 de octubre de 2001. Reingresó con diagnóstico de anemia carencial del lactante prematuro de posible causa ferripriva con Hb 68 g/l, palidez cutáneomucosa y peso 3400g hidratado, no edemas, no dificultad respiratoria, murmullo vesicular normal, no estertores frecuencia respiratoria 52 x min., tonos cardíacos rítmicos bien golpeados, no soplos, no cianosis, buenos pulsos, no gradiente térmico, frecuencia cardiaca 148 x min., no visceromegalias a la palpación, no íctero, lactancia materna exclusiva. Como fue transfundido no se realizaron estudios hasta el próximo ingreso.
Tercer ingreso: 9 de noviembre de 2001, peso 3300 g, palidez cutáneomucosa, hidratado, no edemas, no dificultad respiratoria, murmullo vesicular normal, no estertores, frecuencia respiratoria 52 x min. Tonos cardíacos rítmicos bien golpeados, no soplos, no cianosis, buenos pulsos, no gradiente térmico, frecuencia cardíaca 148 x min. No visceromegalias a la palpación, no íctero, lactancia materna exclusiva.
Ante esta tórpida evolución (curva de peso descendente) y no tener estudios de la anemia se decidió su ingreso en el Servicio de Nutrición
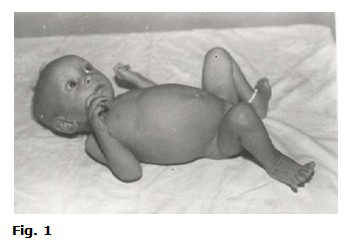
Durante su estadía en el hospital el niño comenzó a presentar deterioro clínico, bioquímico y antropométrico (fig.1 y fig.2).
Se sospechó la FQ y se realizaron estudios genéticos y en espera de los resultados se comenzó con el tratamiento.

Tratamiento impuesto
Niño
Dieta: Lactancia materna más 2 onzas de leche evaporada.
Enzipam o Pancreatina ante de todas las tomas.
Multivitaminas ½ tab al día.
Ácido fólico 1mg al día.
Sulfato de Zinc 3mg al día.
Vitamina C 60mg al día.
Vitamina E 25mg al día.
Aceite en piel.
Sol diario.
A la madre que lacta
Multivitaminas 1tab al día.
Ácido fólico 1mg al día.
Sulfato de Zinc 15mg al día.
Vitamina C 500 mg al día.
Vitamina E 50 mg al día.
Evaluación nutricional clínica
Palidez cutáneomucosa (cara pálida).
Pérdida del panículo adiposo.
Abdomen globuloso.
Pelo ralo y quebradizo.
Mirada triste.
Edema en miembros inferiores.
Dietética
Lactancia materna exclusiva.
Bioquímica
Hb al ingreso 79 g/l
Hb postransfusional 87 g/l
HB al reingreso 68 g/l
Hb postransfusional 103 g/l
Hb 82 g/l.
Proteínas totales 50 g/l
Albúmina 27 g/l
Conteo reticulocitos 15 x 10°
-. CHBCM 240 g/l
-. Lámina periferia
-. Hipocromia ++
-. Anisocitosis +
-. Leucocitos ligeramente aumentados
Creatinina 56 mg
Electrólitos del sudor 176 meq / l de Na.
Resistencia globular normal.
HbF 0, 2%
Electroforesis Hb AA
Evaluación antropométrica
Peso al nacer 6.4 libras referido 36 semanas de gestación.
Curva de peso en descenso.
3450
3400
3300
Talla: 53 cm
Peso: 3300
EP: 3 y 10
ET: 3 y 10
PT – 3p
Resultado del estudio genético
Mutación mayor Df508 / Df508 de la fibrosis quística.
Resultados: 20 de diciembre del 2001.
Actualmente evoluciona con recaídas pero se mantiene estable.
No esta colonizado con pseudomona aureginosa.
DISCUSIÓN
La fibrosis quística, es una enfermedad compleja y es la afección genética de herencia recesiva más frecuente en la raza blanca, causada por una alteración de la proteína (CFTR) que controla el transporte a través de la membrana de las células que segregan moco y que es regulada por un gen localizado en el brazo largo del cromosoma 7. 1, 2
Cuando se sospecha que un paciente puede presentar esta enfermedad, se lleva cabo una serie de estudios que permiten definir la situación en que se encuentra el enfermo desde varios puntos de vistas; en este caso el aspecto nutricional fue el que sugirió el diagnóstico.
La alteración de la nutrición es uno de los graves problemas que afectan a estos pacientes, contribuyen a su deterioro y limitan la supervivencia.
Es también multifactorial y sus mecanismos fisiopatológicos se sustentan en tres pilares fundamentales: déficit de ingesta, aumento de las pérdidas por alteración de los mecanismos de absorción y aumento del gesto metabólico. 3, 4
El déficit de ingesta se debe a anorexia, sobre todo de las infecciones, vómitos por reflujo gastroesofágico, esofagitis y restricciones atrogénicas de la dieta. 5, 6
En relación con el aumento de las pérdidas, la malabsorción debida a la insuficiencia pancreática con aumento del gasto energético por la esteatorrea, pérdidas de heces y sales biliares, micronutrientes y vitaminas, forman parte de muchos sistemas enzimáticos, entre ellos el hierro, como sucedió en este enfermo.
Con el aumento del gasto energético algunos estudios lo han relacionado con los homocigóticos para la mutación Df508, lo cual se demostró en este caso. 7-9
Se conoce que existen genes modificadores, así como el defecto celular al que da lugar la mutación (CFTR) que constituyen factores intrínsicos que ponen en marcha los mecanismos fisiopatogénicos de la malnutrición en portadores homocigóticos. 7, 10
Nuestro paciente presentó una anemia 68g/l sin otras manifestaciones en su primer ingreso, no frecuentes dentro de las manifestaciones nutricionales de la enfermedad, pero pueden estar presentes por los mecanismos expresados anteriormente. Esta forma inusual de presentación impidió la sospecha de esta enfermedad en sus primeros ingresos, hasta que la presencia de una curva de peso en descenso y la aparición de edemas en miembros inferiores sin proceso infeccioso alguno, sugirió la presencia de este diagnóstico como parte de la triada que acompaña a esta enfermedad.
REFERENCIAS BIBLIOGRÁFÍCAS
1. Waldo E. Fibrosis quística. La Habana: Editorial Ciencias Médicas; 1999.
2. Littelewood J. A the south African Cystic Fibrosis Consensus document. A method of management for Cystic Fibrosis in children and adults. World Health Organizarion. 1999.
3. Larriba Bartolomé S. La genética en el Congreso de Denver. Rev Fed Española contr Fibr Quíst. 1999;(27):36.
4. Segal E. Consenso de fibrosis quística. Arch Argent Pediatr. 1999;97(3):188-224.
5. Rojo CM. I Taller Internacional de Fibrosis Quística. Programa nacional de fibrosis quística. Lineamientos generales. 2000.
6. Infante D, Monz J, Lambruschins N, Brusoño C, Clavete JF, Nodal JM, et al. Fibrosis quística: actualización y control en la nutrición pediátrica. Rev Fed Española Contr Fibr Quíst. 2001;(Supl 1):1-32.
7. Dodge A, Jhon E. Guidelines for the diagnosis and managements of Cystic Fibrosis. World Health Organizarion. 1996.
8. Vergara P. Protocolos de fisioterapia respiratoria aplicados a la mucoviscidosis. Rev Fed Española contr Fibr Quíst. 1996;(18):28.
9. Lama More R. Manejo nutricional del niño con fibrosis quística. Rev Fed Española Contr Fibr Quíst. 1998;5(24):36-7.
10. Carugno P, Puig M. La nutrición el lactante preescolar, escolar y adolescente. Méd Inter. 2000;19(3):116-121.
Recibido: 24 de noviembre de 2003.
Aceptado: 21 de abril de 2004.
Dra. Olga Lidia García Peña. Especialista I Grado en Pediatría. Máster en Nutrición. Hospital Provincial Pediátrico Docente Eduardo Agramonte Piña. Camagüey, Cuba.

