










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkEl linfoma de Burkitt (LB) es un linfoma no Hodgkin de células B altamente agresivo.1 Este ha sido denominado por la mayoría de los investigadores que han hecho referencia al tema como el tumor humano de crecimiento más rápido, capaz de lograr una tasa de duplicación celular entre 24 y 26 horas.2,3,4) Es bien conocido que en dicha enfermedad linfoproliferativa se reconocen tres variantes clínicas: el asociado a la inmunodeficiencia, el esporádico (propio de América) y el endémico propio de África ecuatorial.2 Sus extensos estudios epidemiológicos establecieron que esta neoplasia se presenta con mayor frecuencia en niños, adolescentes y adultos jóvenes, ya que es muy rara su aparición en mayores de 40 años.1,5) En el adulto, su desarrollo se asocia, casi invariablemente, al virus de inmunodeficiencia humana (VIH), pues es meritorio de relevancia su diagnóstico en pacientes inmunocompetentes.1,2
La literatura nacional referente al tema es escasa y se basa en informes aislados de casos clínicos. Por tal motivo, no se cuenta con datos que reflejen la magnitud de este problema en el contexto cubano.4,11,12,13 En este artículo se presenta un paciente adulto, inmunocompetente, con diagnóstico de linfoma de Burkitt, cuya forma clínica de debut fue una rotura esplénica espontánea.
Presentación del paciente
Se presenta un paciente masculino de 48 años de edad, con antecedentes de salud aparente, que comenzó, un mes previo a su ingreso, con febrículas vespertinas, pérdida de peso de aproximadamente 7 Kg, y distención abdominal que empeoró progresivamente. Al momento de su ingreso presentó al examen físico una hepatomegalia que rebasaba el reborde costal 5 cm, y gran esplenomegalia que alcanzaba la fosa ilíaca izquierda y pasaba la línea media umbilical. Se le realizaron estudios paraclínicos de urgencia que resultaron en una anemia ligera de 10 g/L normocítica-normocrómica, ligera leucopenia y trombocitopenia. Se realizó un medulograma y una biopsia de médula ósea (BMO) como parte del estudio de un síndrome hepato esplénico con pancitopenia. Se informó la posibilidad de un proceso linfoproliferativo que recordaba a linfoma de Burkitt por su morfología, pendiente de diagnóstico definitivo por BMO. En las primeras 36 horas de estancia hospitalaria el paciente comenzó con: decaimiento, hipotensión, taquicardia, frialdad distal y dolor abdominal generalizado con reacción peritoneal, y caída de la hemoglobina a 4 g/L, en el curso de la rotura esplénica espontánea. El paciente fue intervenido quirúrgicamente y se le realizó una esplenectomía terapéutica y diagnóstica, con biopsia del bazo extirpado. Posteriormente, existió un marcado y rápido deterioro del estado general, con dehiscencia de la sutura abdominal y evisceración, lo cual llevó a la defunción del paciente.
Se le realizaron varios estudios complementarios durante el ingreso hospitalario, que tuvieron los siguientes resultados:
Hemograma: Hb: 4 g/L, VGM: 92 fl, HCM: 29, conteo de leucocitos: 2,5 x 109/L, linfocitos: 0,28, monocitos: 0,02, segmentados: 0,70, plaquetas: 90 x 109/L, reticulocitos: 37 x 10-3. En el coagulograma se observaron: tiempo de sangramiento: 1 minuto, tiempo de coagulación: 9 minutos, tiempo de protrombina: C-13” P-15”, y tiempo de tromboplastina parcial activado: C-30” P-35”. En la lámina periférica se observaron hematíes: normocitosis-normocromía, leucocitos: leucopenia ligera con diferencial adecuado a predominio de granulocitos maduros, plaquetas: ligera trombocitopenia. En la química sanguínea: glicemia: 5,2 mmol/L, creatinina: 64 umol/L, ácido úrico: 306 umol/L, ASAT: 42 umol/L, ALAT: 45 umol/L, GGT: 83 u/L, albúmina: 38 g/L, bilirrubina total: 8 mmol/L, LDH: 960 U/L, proteína C reactiva: 30 U/L, calcio sérico: 2,4 mmol/L. Se le realizaron varios estudios virológicos: VIH (ELISA): negativo, serología VDRL: negativo, Ag de superficie para virus B de la hepatitis: negativo, Ac para virus C de la hepatitis: negativo, y virus de Epstein-Barr: IgM negativo-IgG positivo.
Se le realizó un ultrasonido abdominal en el que se observó un marcado aumento del tamaño del bazo, que medía aproximadamente 20,1 x 8 cm, sin nódulos. Se visualizó un hígado que rebasaba el reborde costal 6 cm, de textura homogénea, sin nódulos, y no se observaron adenopatías.
En el medulograma (Figura 1) se observó la médula ósea hipercelular, con depresión de los sistemas megacariopoyético, eritropoyético y granulopoyético, infiltrada por células linfoides, con núcleo redondeado y citoplasma abundante, basófilo, y con numerosas vacuolas, lo que recordaba el linfoma de Burkitt.
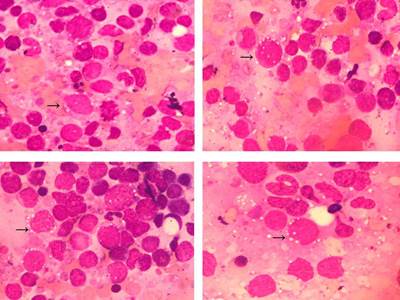
Figura 1 Médula ósea (100x). Tinción May Grumwald-Giemsa. Infiltración por células linfoides de gran tamaño, núcleo redondeado, citoplasma basófilo con numerosas vacuolas
En la biopsia del bazo (Figura 2A) se pudo observar: pérdida de la arquitectura ganglionar por infiltrado masivo de células linfocitarias, aumento del índice mitótico, y abundantes áreas de necrosis, lo que fue concluyente con linfoma B de alto grado de agresividad.

Figura 2.A Biopsia esplénica (100x) Infiltración linfocitaria y pérdida de la arquitectura esplénica normal, grandes áreas de necrosis, numerosas mitosis. B. Biopsia de médula ósea (100x) Infiltración linfocitaria masiva, gran cantidad de células en mitosis. C. Técnica de inmunohistoquímica (400x) CD 20 positivo. D. Ki 67 mayor del 90 %, lo que es propio de linfoma altamente agresivo
En la BMO (Figura 2B) se pudo observar cilindro de médula ósea útil, con más de 6 espacios medulares, infiltrada por células linfocitarias no hendidas, de pequeño y mediano tamaño, y citoplasma basófilo con abundantes vacuolas citoplasmáticas.
En la IHQ (Figura 2C, 2D) se evidenció la positividad para HLA-DR, CD 20, CD10, CD43, IgM y negatividad de CD 5 y Bcl 2. Ki 67 98 %.
Comentario
Los linfomas no Hodgkin (LNH) constituyen un grupo importante y heterogéneo de enfermedades linfoproliferativas, dentro de las cuales, el LB representa menos del 1 % de los casos, con una incidencia en adultos de 2,5 casos por millón de habitantes por año.1,3,6,14
El caso informado se trata de una forma esporádica de la enfermedad. Esta se presenta típicamente en adultos menores de 35 años, con predominio en sexo masculino de 4:1, compromiso abdominal, y frecuentemente, infiltración ósea (30 %), sistema nervioso central (SNC) (15 %), y también renal, testicular, ovárico, mamario o de médula ósea.7,8) A pesar de que la afectación intrabdominal con compromiso visceral es lo más común en estos pacientes, no se encontraron estudios previos en la literatura revisada que referenciaran la rotura esplénica como complicación al debut de la enfermedad. La mayoría de los informes de caso se limitaban a la afectación abdominal y hacían referencia a una enfermedad que requería laparotomía por obstrucción intestinal por masa abdominal, por invaginación, o por «abdomen agudo», debido a una pseudoobstrucción intestinal.5
La presentación clínica se caracteriza por el crecimiento rápido de masas, presencia de síntomas B (fiebre, pérdida de peso y sudoraciones nocturnas profusas), y a menudo, evidencia clínica de lisis tumoral espontánea. 7 Por otra parte, en menos del 10% hay un compromiso de la médula ósea, lo cual hace aún más distintivo el actual estudio.1
Por varias razones esta afección ha sido considerada un enigma en la investigación del cáncer. En parte, por su particular distribución geografía que se acompaña no solo por diferencias en su incidencia, sino por sus características clínicas, y de forma relevante, por su asociación con la infección por el virus de Epstein-Barr (EBV). Esta compleja relación causal entre EBV y LB no ha sido aclarada luego de 40 años desde su descripción como carcinógeno.9 La infección por EBV se observa en el 25-40% de estos casos, y sobre todo, asociado a la forma endémica de la enfermedad; no obstante, se puede ver en cualquiera de sus variantes, como ocurre en el presente caso.1
Histológicamente, el LB presenta una infiltración monomórfica, de células no hendidas de mediano tamaño, con núcleo redondeado y citoplasma basófilo, con vacuolas lipídicas citoplasmáticas prominentes. Tiene alta tasa de proliferación con Ki67+ 100 %, en la generalidad de los casos. Las células de Burkitt producen un patrón difuso de compromiso tisular que bajo el microscopio se caracteriza por la presencia de una apariencia de «cielo estrellado», también observado en otros linfomas altamente proliferativos, con macrófagos dispersos con restos nucleares en su interior.4,7,14
Expresan IgM de superficie y antígenos asociados a la estirpe B (CD19, 20, 22, 79a), así como CD10, HLA-DR y CD 43, ausencia de CD5, BCL-2, TdT y típicamente CD23.7,10
El LB es un tumor altamente agresivo, pero con muy buena respuesta al tratamiento con altas dosis de quimioterapia, por lo que el diagnóstico precoz y el tratamiento inmediato (dentro de las 48 horas que siguen al diagnóstico) son indispensables.7,8 En el presente caso, a pesar de que el diagnóstico se realizó tras un breve período de tiempo después de su ingreso, el desarrollo de una complicación con necesidad de intervención quirúrgica de urgencia, constituyó un factor que retrasó el inicio del tratamiento específico, lo que influyó directamente en la evolución desfavorable hasta el desenlace fatal.
En esta publicación se presentó un caso donde la falta de tratamiento y sospecha diagnóstica oportuna llevaron a un rápido deterioro del paciente, que propició una mala evolución clínica. En el contexto cubano, el LB no tiene una incidencia frecuente, por lo que cada informe de caso que se realice constituye un medio para comunicar información científica sobre esta neoplasia linfoide que requiere un rápido diagnóstico.

