










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La hemofilia es una enfermedad con herencia mendeliana que se transmite a través de un patrón recesivo ligado al cromosoma X. Se produce por mutaciones a nivel de los genes del factor VIII de la coagulación (Hemofilia A), localizado en Xq28 y factor IX (Hemofilia B) con locus en Xq27. Como consecuencia se produce una disminución o deficiencia funcional de estas proteínas en el plasma, por lo que clínicamente ambas condiciones se expresan como trastornos hemorrágicos congénitos.1,2
La enfermedad es heredada en el 70 % de los casos, mientras que el 30 % restante se presenta como consecuencia de una nueva mutación en individuos sin antecedentes familiares. Se plantea que estos casos esporádicos pudieran deberse a “mutaciones de novo” que ocurren durante los procesos del ADN del nuevo organismo en desarrollo o en menor medida a mutaciones heredadas de la madre debido a mosaicismo gonadal, o sea, daños en el ADN materno confinados solo a células germinales. Sin embargo, en la mayoría de los casos la condición es heredada de una madre portadora que a su vez recibe la mutación ocurrida durante la gametogénesis de su progenitor masculino.1,2
Para la hemofilia A, particularmente existe una gran heterogeneidad de defectos moleculares, que incluyen mutaciones puntuales, mutaciones en el sitio de empalme, grandes deleciones, pequeñas deleciones e inserciones que generan cambios en el marco de lectura, así como inversiones.3 Estas alteraciones moleculares presentan una correlación con la severidad del fenotipo (leve, moderado y grave), al ser más frecuentes en un fenotipo que en otros.4
En el caso de la hemofilia B, las mutaciones de sentido erróneo son los cambios moleculares más frecuentes (65 %), además se describen mutaciones sin sentido y en el sitio de empalme. Otras variaciones corresponden con deleciones, inserciones y duplicaciones. Se ha descrito que la mayor prevalencia de mutaciones de sentido erróneo en la hemofilia B comparado con la hemofilia A, puede ser uno de los posibles factores que contribuye a que el fenotipo clínico sea más moderado en los pacientes con hemofilia B grave.2,5
Para las familias con antecedentes de hemofilia, resulta de suma importancia la identificación de las mujeres portadoras, lo cual permite desarrollar procesos de asesoramiento incluso antes de que estas féminas decidan tener descendencia. 6 Antes de proceder a estudios de ADN se requiere identificar y clasificar las portadoras de acuerdo a los criterios establecidos:
Portadoras obligadas6
· Todas las hijas de un varón enfermo (reciben el único cromosoma X con la mutación de su padre).
· Mujeres con un hijo varón enfermo y antecedente de varones hemofílicos por vía materna (tíos, hermanos, etc.)
· Mujeres con más de un hijo hemofílico de embarazos independientes o gestaciones gemelares dicigóticas.
Portadoras posibles6
· Mujeres con un solo hijo hemofílico y sin antecedentes familiares.
· Todas las hijas de una mujer portadora obligada.
· Todas las mujeres con antecedentes familiares de hemofilia por línea materna.
El desarrollo y aplicación de las técnicas de biología molecular ha permitido una mejor caracterización de los genes codificantes de los factores de coagulación VIII y IX. El diagnóstico molecular de la hemofilia se realiza por medio de estudios directos, los cuales a través de múltiples técnicas logran identificar la mutación causante de la enfermedad y correlacionarla con la severidad del fenotipo.7 Otro método, sin los beneficios antes descritos pero que no deja de ser valioso es el análisis indirecto de ADN, basado en la identificación de polimorfismos del gen, útil en el diagnóstico de portadoras.7,8
Cuba no dispone hasta el momento, de métodos directos para el diagnóstico de hemofilia, lo que limita la caracterización de cada paciente afectado y con ello el manejo individualizado del enfermo y su familia. No obstante, existe la posibilidad de estudio indirecto familiar para diagnóstico de portadoras, lo que ofrece opciones de diagnóstico prenatal en futuras gestaciones.
Pinar del Río registra actualmente 10 pacientes hemofílicos en edad pediátrica, cuyas familias no están totalmente estudiadas desde el punto de vista molecular, lo que limita el manejo del riesgo de recurrencia y los procesos de asesoramiento genético. Ante esta panorámica se realiza la presente investigación que resume los resultados de estudios moleculares indirectos realizados en familias pinareñas con antecedentes de hemofilia.
MÉTODOS
Se realizó un estudio descriptivo, observacional y transversal con el universo de los 10 pacientes hemofílicos en edad pediátrica procedentes de Pinar del Rio y sus familias. La muestra estuvo conformada por cinco familias a quienes se realizó estudio molecular indirecto para diagnóstico de portadoras y consecuentemente diagnóstico prenatal, luego del requerido asesoramiento genético y el consentimiento por parte de los participantes en el estudio.
Las muestras obtenidas (sangre y líquido amniótico según el caso) se procesaron en el laboratorio de Biología Molecular del Centro Nacional de Genética Médica. Para Hemofilia A se utilizaron cuatro marcadores polimórficos: St14, BcLI, HindIII y Xba. Para Hemofilia B los marcadores usados fueron Ddel, HhaI, TaqI y MnlI.
Se cumplió con los principios de la ética médica y los aspectos establecidos en la Declaración de Helsinki.
RESULTADOS
Del universo de 10 pacientes varones menores de 18 años en Pinar del Rio, nueve (90 %) tuvieron diagnóstico clínico y bioquímico de hemofilia A. Por tanto, uno solo (10 %) se diagnosticó como déficit de factor IX. (Hemofilia B)
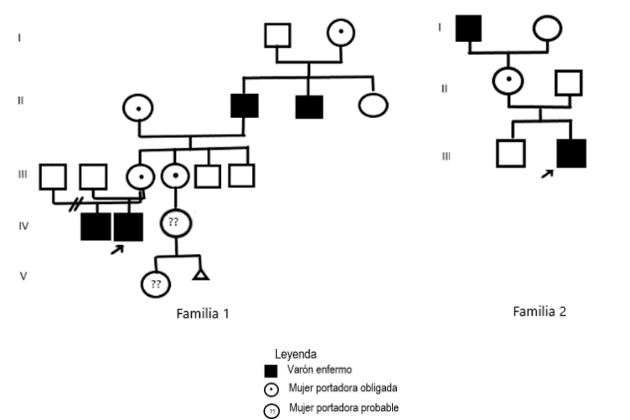
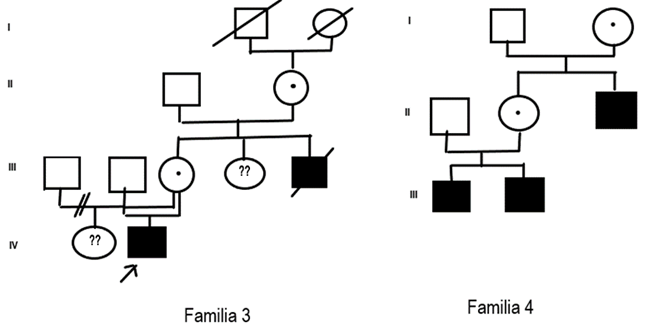
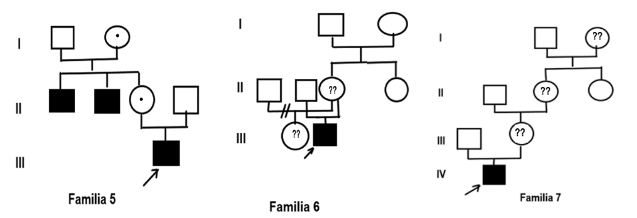
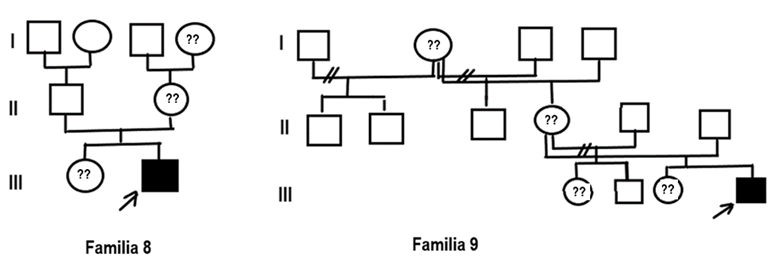
Del total de la muestra, el 60 % presentaron antecedentes familiares de Hemofilia al momento del diagnóstico y el resto (40 %) se diagnosticaron como casos nuevos (Fig. 1, Fig. 2, Fig. 3 y Fig. 4).
Ninguna de las nueve familias refirió antecedentes de consanguinidad conocida, al menos en las cuatro generaciones anteriores.
Para Hemofilia A, el marcador más informativo fue St14 (75 %), seguido de BcLI (50 %) y HindIII (25 %). Con ellos fue posible diagnosticar siete mujeres portadoras (evaluadas como probables antes del estudio) (familia 1, familia 4, familia 6 y familia 7) y se confirmó una portadora obligada (familia 5). Se procedió al diagnóstico prenatal de un varón enfermo y uno sano. En el embarazo con feto afectado la familia decidió continuar con la gestación. Resta un tercer feto estudiado pendiente de resultado.
Luego del análisis de cada genealogía se decidió que la familia 2 no requería de estudio molecular por el momento. La única mujer es portadora obligada y ha concluido su reproducción. Quedan pendientes los estudios de portadoras de las familias 8 y 9 para hemofilia A.
Para Hemofilia B (familia 5) la única familia incluida en la muestra resultó informativa para los marcadores Ddel y HhaI. Se diagnosticó una mujer portadora y un feto enfermo. La pareja optó por la continuidad del embarazo.
DISCUSIÓN
Tanto la hemofilia A como la variante B constituyen trastornos hemorrágicos, en su mayoría congénitos, que afecta mayoritariamente a los hombres, dada su transmisión según un patrón recesivo ligado al cromosoma X.1,2 Sin embargo, se reportan, aunque en menor proporción, mujeres con sintomatología hemorrágica debido a déficit de factores de la coagulación.9 En el estudio con familias pinareñas que conviven con hemofilia la totalidad de los afectados sintomáticos fueron varones como se reporta en casi la totalidad de los estudios poblacionales que abordan este desorden.10,11,12
Los resultados con relación a la presencia de historia familiar positiva de hemofilia son consistentes con la literatura. Más de la mitad de los pacientes tienen un pariente afectado que se puede reconocer con un modelo de segregación mendeliana.1,2,3,8 Los casos esporádicos, pudieran ser resultado de mutaciones de novo en el cigoto del nuevo individuo, o bien hijos de portadoras en las que se originó la nueva mutación.13 Otros mecanismos como el mosaicismo gonadal o la transmisión de una mutación en heterocigosis de una generación de mujeres a otra sin que ocurra el nacimiento de varones enfermos en una familia, también se reportan.1,2
Pudiera ser el caso de la familia 7, en la que sin antecedentes familiares conocidos se logra identificar una madre portadora.
Para hemofilia A el marcador polimórfico más informativo resultó el St14. Cabe señalar que son escasas las referencias recientes sobre el uso de este marcador extragénico en el diagnóstico de portadoras de Hemofilia. Se trata de una tecnología del siglo XX, que aun cuando se utiliza, ha dado paso progresivamente a otros métodos de análisis.8 En estudios realizados en Cuba anteriormente en familias con hemofilia procedentes de todo el país, se describen índices de informatividad similares o discretamente más bajos.4,14,15
Por otra parte, el marcador intragénico BcLI fue informativo en dos familias y el HindIII solo en una, datos inferiores a lo reportado por otros autores dentro y fuera de Cuba.4,8,14 Aunque la muestra es escasa, pudiera inferirse que la tasa de heterocigocidad para estos marcadores es baja en población pinareña. Ello sugiere extender el estudio a las familias pendientes de análisis y diseñar análisis de las frecuencias alélicas para estos polimorfismos según las diferentes regiones del país. No obstante, hasta el momento ha sido significativa la utilidad del método aplicado en el diagnóstico de portadoras de Hemofilia A, aun cuando sería conveniente ampliar el número y la diversidad de los marcadores a utilizar.
Para la hemofilia B, la única familia estudiada resultó informativa para los marcadores Ddel y HhaI. Aunque fue útil en este caso, una sola genealogía no es suficiente para validar el uso de estos polimorfismos, cuyas referencias, como sucede con la Hemofilia A, se remontan a las últimas décadas del siglo pasado. Uno de los estudios más cercanos en el tiempo reporta la notable variación en la capacidad informativa de estos marcadores RFLP vinculados al locus de la hemofilia B en grupos de población de la India lo que implica modificaciones en la estrategia para el diagnóstico de portadoras, lo que sugiere una selección prudente de los marcadores en función de grupos de población específicos, así como el uso de marcadores adicionales.15
De manera general, aunque la caracterización molecular por métodos directos es el estándar de oro en el diagnóstico de la hemofilia, constituye un verdadero desafío en países con limitados recursos. Por tanto, el análisis indirecto de marcadores de ADN vinculados a los loci de FVIII y FIX, se presenta como una alternativa más práctica para la detección de portadoras y el abordaje familiar, que aporta elementos para diagnóstico, asesoramiento y manejo del riesgo de recurrencia de la hemofilia en cada genealogía de manera particular.

