










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
En el desarrollo de la forunculosis recidivante se han considerado múltiples factores de riesgo, entre los que se encuentran factores genéticos como los antígenos de histocompatibilidad HLA-DR3 (del inglés Human Leucocyte Antigens) que predisponen a la portación nasal de Staphylococcus aureus -S. aureus-,1 los polimorfismos genéticos en genes de receptores de glucocorticoides humanos y los receptores de interleucina-4 (IL-4) (genotipo C524T)2 y de IL-6.3
En muchos adultos jóvenes con forunculosis recidivante no se detecta la presencia de factores ambientales y el contagio endógeno es más frecuente que el exógeno. Debe tenerse en cuenta que aproximadamente el 30% de los adultos están colonizados por S. aureus; con mayor frecuencia en la mucosa nasal.4
La susceptibilidad genética a las infecciones muestra que los diferentes componentes genéticos juegan un papel importante en la determinación diferencial de la susceptibilidad a las principales enfermedades infecciosas de los humanos. La genética epidemiológica y los estudios de gemelos proporcionan evidencia significativa con respecto a la variación genética en las poblaciones humanas porque contribuye a la susceptibilidad a esas enfermedades. La genética humana de las enfermedades infecciosas ha postulado que un raro grupo de inmunodeficiencias primarias confiere vulnerabilidad a múltiples enfermedades infecciosas (un gen, múltiples infecciones), mientras que las enfermedades infecciosas comunes están asociadas con la herencia poligénica de múltiples genes de susceptibilidad (una infección, múltiples genes). Simultáneamente, se ha determinado, en varias infecciones comunes, la herencia de un gen principal de susceptibilidad, al menos en algunas poblaciones. Este nuevo paradigma (un gen, una infección) desdibuja la distinción entre la genética mendeliana basada en pacientes y la genética de enfermedades complejas basadas en estudios de población, que determina una nueva forma de abordaje conceptual para explorar las bases moleculares de la genética de enfermedades infecciosas en los humanos, las que tienen una fuerte influencia epigenética.5,6
Bajo esta perspectiva se desarrolló una investigación (algunos de sus resultados se divulgan en esta publicación) con el objetivo de identificar la susceptibilidad de base genética en individuos afectados por forunculosis recidivante según la agregación familiar verdadera de la enfermedad en un grupo de familias con individuos afectados procedentes de cinco municipios de la Provincia de Villa Clara.
MÉTODOS
Se realizó un estudio analítico transversal con el empleo de un diseño de estrategia familiar correspondiente al campo de la epidemiologia genética. Se analizó la información obtenida a través de las genealogías para identificar evidencias de susceptibilidad genética y se empleó, específicamente, el análisis de la agregación familiar verdadera.
A través de la obtención del árbol genealógico de la familia conformaron las cohortes de todos los familiares de los integrantes, lo que permitió considerar diferentes antecedentes genéticos y establecer agregación familiar verdadera para la totalidad de los parientes, por grado de parentesco y para parientes específicos.
Los árboles genealógicos fueron realizados hasta la cuarta generación por la metodología de la Escuela francesa, de uso común en Cuba para dos grupos.7
Para la reconstrucción de las dos cohortes del primer grupo empleadas en la investigación, los 132 individuos enfermos estudiados se dividieron en dos grupos en base a la edad, debido a que en el grupo de 30 individuos enfermos de tres a 17 años, entre los parientes de primer grado, no se podían incluir hijos y, en los de segundo grado, ni sobrinos ni nietos; en la cohorte de 102 individuos de 18 años y más se incluyeron todos los tipos de parientes específicos. Por la misma razón en la cohorte del segundo grupo, cuando se compara con la cohorte de tres a 17 años, se excluyeron hijos, sobrinos y nietos, mientras se utilizaron la totalidad de los parientes para la comparación con la cohorte de familiares de individuos de 18 años y más.
El análisis de agregación familiar verdadera se llevó a cabo mediante la reconstrucción de dos cohortes. Las cohortes de familiares del primer grupo quedaron constituidas por 2 281 parientes totales en 102 genealogías de 18 años o más y 487 parientes totales en 30 genealogías entre tres y 17 años. Las cohortes de familiares del segundo grupo se conformaron con 1 102 parientes totales en 43 genealogías y 933 parientes empleados para el análisis en el grupo de tres a 17 años, después de excluir los hijos, los sobrinos y los nietos.
Para el cálculo de riesgo por agregación familiar verdadera para tipos específicos de parientes la muestra total del primer grupo fueron 132 individuos, 30 en el grupo entre tres y 17 años y 102 en el grupo de estudio de 18 años y más, respecto a 43 del segundo grupo, y se muestran los resultados para los cinco tipos de parientes específicos que pudieron analizarse tanto en la cohorte de familiares del primer grupo de tres a 17 años, como en la de 18 años y más.
Para el cálculo de riesgo por agregación familiar verdadera para tipos específicos de parientes la muestra total del segundo grupo fueron 132 individuos, 30 en el grupo entre tres y 17 años y 102 en el grupo de estudio de 18 años y más, respecto a 43 del segundo grupo.
Agregación familiar verdadera: variable compleja, determinada cuando la aglomeración de casos en una familia es superior a lo esperado por el azar. Se utilizaron dos de los tres criterios establecidos:
Criterio 2: se definió que había agregación familiar si la prevalencia entre los parientes del caso era mayor que la prevalencia entre los parientes del control. Las variables simples empleadas para el cálculo de la agregación familiar verdadera por criterio 2 fueron:
En los grupos de estudio:
Familiar de primer grado afectado (madre, padre, hermano, hijos)
Familiar de segundo grado afectado (tíos, sobrinos, abuelos, nietos, medios hermanos)
Familiar de tercer grado afectado (primos)
Total de familiares de cualquier grado afectados
Parientes totales por grado de parentesco
Parientes totales
Criterio 3: se definió que había agregación familiar si dentro de los familiares del segundo grupo la prevalencia entre parientes de primer grado era mayor que la prevalencia entre parientes de segundo y tercer grados de conjunto.
Variables simples empleadas para el cálculo de la agregación familiar verdadera por criterio 3.
En el segundo grupo de estudio:
Familiar de primer grado afectado
Total de familiares de primer grado afectados o no
Familiar de segundo y tercer grado afectados
Total de familiares de segundo y tercer grados afectados o no.
La evaluación de riesgo por agregación familiar en la forunculosis recidivante por tipos específicos de parientes se hizo de la siguiente forma: se construyeron tablas de contingencia de 2x2l, se analizaron por separado el grupo de estudio entre tres y 17 años y el grupo de 18 años y más y se hicieron evaluaciones de riesgo por separado para la existencia exclusiva de antecedente genético de los siguientes parientes, sin considerar el sexo: progenitores, hermanos, tíos, abuelos y primos afectados (ubicados en la celda Si); de no existir estos antecedentes los valores fueron ubicados en la celda No. Se procedió de igual forma para las celdas correspondientes a los controles.
Se estudió la agregación familiar mediante los criterios 2 y 3 para dos situaciones: se consideró la totalidad de parientes y para familiares por grado de parentesco en general se realizaron agrupaciones para parientes de primero, segundo y tercer grados, de la forma establecida, por el por ciento de genes en común entre parientes.8
Se determinó la proporción de parientes afectados para parientes totales y por grado de parentesco y compararon mediante la prueba de hipótesis de homogeneidad de Chi cuadrado.
Para analizar el riesgo por agregación familiar para forunculosis recidivante se hizo el análisis para cinco tipos de parientes específicos. Se emplearon tablas de contingencia bivariadas, calculó la significación mediante el estadígrafo de Chi cuadrado de la prueba de independencia. Cuando existieron diferencias significativas se calculó la razón de productos cruzados (OR, del inglés Odds Ratio) y el intervalo de confianza al 95%. La fuerza de la asociación se evaluó mediante la V de Cramer.
Para todas las pruebas de hipótesis se prefijó un valor de significación alfa de 0,05 para la toma de la decisión estadística.
Se solicitó el consentimiento informado, por escrito, de los adultos participantes o de los padres de los niños incluidos.
RESULTADOS
La Tabla 1 resume la agregación familiar por los criterios 2 y 3 de la agregación familiar en la cohorte del grupo de estudio entre tres y 17 años. Al analizar el criterio 2 se comprueba que, entre los 487 familiares de cualquier grado del primer grupo entre tres y 17 años, hubo 77 enfermos (15,8% de prevalencia), respecto a 3,10% entre los familiares del segundo grupo (p=0,000). Este resultado también se observó para parientes de primero, segundo y tercer grados por separado, lo que evidencia que hubo agregación familiar por el criterio 2.
Al analizar el criterio 3 también se comprobó que existe agregación familiar porque, dentro del primer grupo, la prevalencia entre parientes de primer grado fue de 40% respecto a 10,7% entre los familiares de segundo y tercer grados de conjunto, diferencias significativas.
Tabla 1 Agregación familiar en la cohorte del grupo de estudio entre tres y 17 años
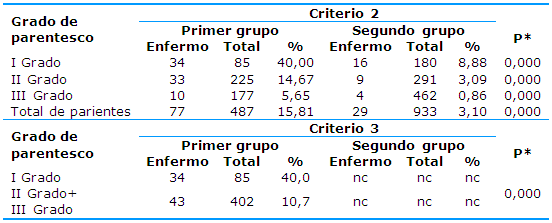
I, II y III Grados: primer, segundo y tercer grados de consanguinidad; nc: no corresponde
*Significación estadística de la prueba de homogeneidad de Chi cuadrado
Los datos se presentan en totales y por cientos; significación p≤0,05
Fuente: Base de datos de las genealogías de las familias
En la tabla 2, al analizar el criterio 2 en la muestra de individuos de 18 años y más se comprueba, para la totalidad de parientes, que la prevalencia entre 2 281 familiares de cualquier grado de los adultos fue de 178 enfermos (7,80% de prevalencia), respecto a 4,45% entre los familiares (p=0,000); lo mismo se observó para parientes de primer y tercer grados, por separado. No hubo diferencias en el comportamiento de los parientes de segundo grado. Este resultado muestra agregación familiar mediante el empleo del criterio 2 para la totalidad de los parientes y para parientes de primer y tercer grados.
Al analizar el criterio 3 también se constató agregación familiar se comprobó que dentro de los familiares del primer grupo la prevalencia entre parientes de primer grado fue de 22,6% y los familiares de segundo y tercer grados, de conjunto, tuvieron una frecuencia de 4,46%, diferencias que resultaron significativas (Tabla 2).
Tabla 2 Agregación familiar en la cohorte del grupo de estudio de 18 años y más
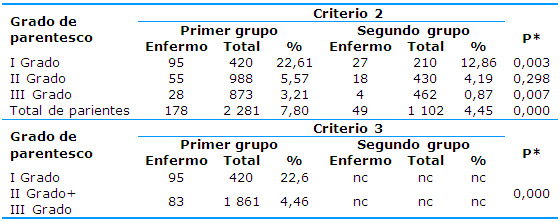
I, II y III Grados: primer, segundo y tercer grados de consanguinidad; nc: no corresponde
*Significación estadística de la prueba de homogeneidad de Chi cuadrado
Los datos se presentan en totales y por cientos; significación p≤0,05
Fuente: Base de datos de las genealogías de las familias
En la cohorte de los familiares del primer grupo entre tres y 17 años (Tabla 3) se evidencia la existencia de riesgo por agregación familiar debido a la existencia de progenitores enfermos (OR=5,82), tíos (OR=5,50), abuelos (OR=10,0) y hermanos (OR=4,18), mientras que no fue comprobable mediante el comportamiento genealógico de los primos (OR=2,89).
Tabla 3 Riesgos por agregación familiar mediante análisis de parientes específicos en la cohorte del grupo de estudio entre tres y 17 años
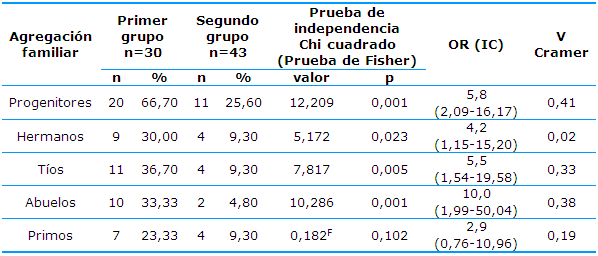
p: significación estadística; OR: Odd Ration; IC: intervalo de confianza
Los datos se presentan en totales y por cientos
Fuente: Base de datos de las genealogías de las familias
En la cohorte de los familiares del primer grupo de 18 años y más (Tabla 4) existe riesgo por agregación familiar; este resultó significativo por la presencia de hermanos enfermos (OR=3,17), pero no fue comprobable mediante el comportamiento genealógico de los progenitores (OR=1,66), tíos (OR=2,04), abuelos (OR=2,17) y primos (OR=1,77).
Tabla 4 Riesgos por agregación familiar mediante análisis de parientes específicos en la cohorte del grupo de estudio de 18 años y más

p: significación estadística; OR: Odd Ration; IC: intervalo de confianza
Los datos se presentan en totales y por cientos
Fuente: Base de datos de las genealogías de las familias
DISCUSIÓN
Las enfermedades multifactoriales tienen un componente hereditario complejo con influencia de fenómenos ambientales, no siguen los patrones clásicos de herencia y se presentan en individuos de una misma familia en diferentes generaciones. Los individuos de una misma familia comparten una mayor proporción de sus genes entre ellos que con individuos no emparentados de la población. Una característica primaria de enfermedades con herencia compleja es que los individuos afectados tienden a agruparse en familias, esta característica se denomina agregación familiar, y suele deberse, con frecuencia, a causas hereditarias, aunque no de manera absoluta, porque individuos de la misma familia comparten hábitos y estilos de vida que implican otros factores de riesgo no genéticos.8
La agregación familiar se demostró para los parientes de primer, segundo y tercer grados de acuerdo con lo que se establece por el criterio 2 en la cohorte de casos entre tres y 17 años; este es un resultado esperado porque los parientes de primer grado (padre, madre y hermanos) comparten el 50% de la información genética. Los de segundo grado (tíos, sobrinos, y abuelos) el 25% y los de tercer grado (primos) comparten el 12,5%.8 Se observaron diferencias en el comportamiento de las cohortes porque la de familiares de individuos de 18 años y más no mostró agregación familiar para parientes de segundo grado, para el resto de los parientes se comportó semejante a los casos entre tres y 17 años. En opinión de los autores no se puede descartar que la información genealógica resulte más cercana en la cohorte de los casos entre tres y 17 años que en la de los pacientes de 18 años y más y tampoco que se le de el mismo nivel de importancia a la información genealógica aportada por los familiares en la entrevista, a pesar de los controles tenidos en cuenta para evitar los sesgos.
Se demostró agregación familiar entre los familiares de los casos de acuerdo con lo establecido en el criterio 3 para ambas cohortes.
Al explicar las posibles causas de la agregación familiar verdadera encontrada hay que referirse a los informes de varios polimorfismos de los genes que determinan el reconocimiento de patógenos, las proteínas solubles como la MBL,9 los receptores que provocan endocitosis (receptores para FcR-γ)10 y los receptores que provocan señales intracelulares TLRs.11,12
De la misma forma se han descrito varios polimorfismos de diferentes interleukinas (IL-1, IL-6 y IL-10) asociados a la infección y a su evolución tórpida en pacientes sometidos a intervenciones quirúrgicas.3,13,14,15
Un polimorfismo de simple nucleótido en la enzima DNA metiltrasferasa-3A (DNMT3A), que resulta en baja actividad de metilación, se correlaciona con persistencia de bacteriemia por S. aureus e incremento de los niveles séricos de IL-10. Un cambio de patrón de expresión genética se correlaciona con protección contra la persistencia bacteriana y bajos niveles del IL-10, en el que los heterocigotos tienen más posibilidades que los homocigotos de resolver la bacteriemia.16
La aparición de la enfermedad dentro de las familias tendría entonces dos direcciones: primero, las de los miembros de la familia que comparten genes ancestrales comunes en los que pueden estar incluidos genes de susceptibilidad o resistencia a la infección por S. aureus y, por tanto, tener un comportamiento diferencial en base a la constitución genética en un ambiente especifico, y segundo, el patrón seguido por los miembros legales de la familia en los que la presentación obedece a contagio ambiental.
Al analizar los resultados del estudio de riesgo por agregación familiar en la cohorte de individuos entre tres y 17 años el riesgo resultante de la medición de la agregación familiar solo por la existencia de algún tipo específico de pariente de primer grado enfermo (progenitores o hermanos) entre todas las genealogías de casos respecto a la existencia de estos mismos parientes enfermos entre todas las genealogías de controles arrojó la existencia de riesgo, al igual que cuando se analizó el comportamiento de la agregación familiar mediante tíos o abuelos (parientes de segundo grado). Este es un resultado esperado para una enfermedad multifactorial debido a que el por ciento de genes comunes del mismo origen ancestral que deben compartir parientes de primer y segundo grados y también sería más probable encontrar, como se observó en los resultados, que este riesgo no se confirmó cuando se analizó el comportamiento de la frecuencia de primos enfermos entre casos y controles.
Sin embargo, teniendo en cuenta que la forunculosis recidivante es una enfermedad infecciosa, y por tanto presumiblemente ambiental, este resultado es realmente novedoso; no obstante, se han encontrado diversas evidencias de la participación de genes en la susceptibilidad o la resistencia a las infecciones humanas.
En la literatura revisada se informan polimorfismos genéticos que determinan predisposición para las infecciones por S. aureus. La mutación en el gen STAT3 afecta la diferenciación de los linfocitos Th17 CD4.17
La presencia del haplotipo de antígenos de histocompatibilidad del sistema HLA-DR3 que predispone a la portación nasal de S. aureus, los polimorfismos genéticos en receptores de IL-4 (genotipo (C524T)18 y la haploinsuficiencia para el gen de OTULIN en el cromosoma 519 son mutaciones que predisponen a tener infecciones por S. aureus. El gen de OTULIN codifica para una proteína que participa en el control de la inflamación y elimina marcas de ubiquitina que se unen a ciertas proteínas en respuesta a la presencia de un agente extraño. Cuando ambas copias del gen están alteradas las rutas de activación de la inflamación están anormalmente activas, lo que deriva en una enfermedad conocida como otulipenia. Las mutaciones detectadas en los pacientes con infección grave por estafilococos inducen la formación de agregados de ubiquitina y acumulación de otra proteína en los fibroblastos de la piel, lo que facilita el daño citotóxico de la toxina liberada por S. aureus.19
Ciertas mutaciones que causan alteraciones en las funciones de las células de Langerhans incrementan las infecciones cutáneas por S. aureus y empeoran la resolución a las mismas. El inflamasoma (NLRP3) en los macrófagos y los neutrófilos es la llave del aclaramiento de S.aureus a nivel cutáneo. Los polimorfismos encontrados en este receptor (Q705K/C10X) pueden disminuir la apoptosis de neutrófilos, lo que lleva a la disminución del aclaramiento intracelular. Además, el polimorfismo (29940G˃C SNPs) detectado en sepsis clínica ha demostrado que deprime la respuesta de citocinas inflamatorias, lo que protege de pobre evolución clínica de la enfermedad.20
A partir de los resultados de estudios experimentales que demostraron asociación del cromosoma 8 murino con la susceptibilidad a las infecciones por S. aureus se encontró 11 genes candidatos que se expresan de manera significativa en humanos infectados, cuatro con alta frecuencia de expresión en neutrófilos retados por antígenos bacterianos (Ier2, Crif1, Cd97 y Lyl1) y dos (Cd97 y Crif1) que juegan un importante papel en las defensas contra S. aureus.21
La agregación familiar que se estudia es referida a una base genética poligénica en la que, presumiblemente, pueden intervenir muchos genes que podrían conferir susceptibilidad o resistencia a la infección por S. aureus; no se trata de mecanismos de segregación alélica simple.
Esos diferentes poligenes, que pueden ser polimorfismos, tienen a su vez una interrelación compleja entre ellos, en la que se han planteado mecanismos aditivos o multiplicativos sobre el efecto fenotípico final.
No está claro si cada poligen o polimorfismo tiene el mismo efecto sobre el fenotipo, más bien, la idea es que los efectos son diferentes, incluso pueden ser de sentido contrario. Estos mecanismos se pueden complejizar aún más en interacción con factores ambientales y el control de su expresión puede estar regulado por mecanismos epigenéticos.6,22
La población cubana se ha formado por un proceso de interacción y mezcla racial entre españoles y negros africanos fuertemente matizado por los procesos migratorios internos y externos.23
Este mestizaje, aunque se plantea como responsable de que en algunos estudios de polimorfismos no se encuentre la asociación informada en otras latitudes, más recientemente se emplea con éxito a través de una técnica denominada mapeo de mezcla que permite encontrar con más rapidez asociaciones de interés.24
El análisis de agregación familiar es un paso inicial importante para caracterizar los determinantes genéticos de los fenotipos en los estudios epidemiológicos. El objetivo del análisis de agregación familiar es identificar grupos de individuos relacionados que presentan los mismos síntomas o las mismas enfermedades debido a algún mecanismo compartido subyacente. Una amplia gama de enfermedades se han analizado previamente con métodos de agregación familiar: el cáncer, las enfermedades cardiovasculares y, recientemente, las enfermedades autoinmunes y el insomnio.25
En relación con las infecciones, y específicamente las producidas por S. aureus, es un análisis novedoso no realizado antes en este contexto y relativamente poco abordado en la literatura internacional. En 2016 se realizó un estudio en Dinamarca26 para evaluar la agregación familiar en la susceptibilidad a S. aureus, pero solo se evaluaron parientes de primer grado y se utilizó el criterio 1 de agregación familiar, en el que se efectuó un análisis de la incidencia observada entre padres y hermanos de individuos que, según el Registro Nacional, habían estado hospitalizados por una infección grave producida por esta bacteria. Con respecto a la incidencia esperada en base a la incidencia poblacional encontraron mayor riesgo que para la población no expuesta a ese antecedente, así como que el riesgo era mayor cuando el enfermo era un hermano, resultado que concuerda con este estudio, en el que solo se demostró riesgo por agregación familiar en la cohorte de adultos cuando se evaluó para parientes aislados en el caso de los hermanos enfermos, no así para los familiares del grupo de estudio de tres a 17 años, en el que se mostró riesgo por agregación familiar para varios tipos de parientes. Los resultados encontrados constituyen un aporte a los esfuerzos por demostrar una contribución de base genética a la variabilidad fenotípica total en las infecciones cutáneas recidivantes producidas por S. aureus.
CONCLUSIONES
La agregación familiar verdadera en la forunculosis recidivante, tanto para la totalidad de los parientes como para grados de parentesco específicos, así como el antecedente de agregación familiar de forunculosis como factor de riesgo, muestra el lugar de la herencia en la susceptibilidad o la resistencia a esta infección en individuos con lazos consanguíneos.

