










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El síndrome Nance Horan (SNH), que incluye dentro de su fenotipo clínico la presencia de cataratas congénitas y anomalías dentales, es un trastorno congénito de origen genético extremadamente raro, al que se le conoce también como síndrome catarata-dental.1 Mediante estudios de ligamiento genético se determinó que el gen del SNH (OMIM 302350) tiene su locus génico en el brazo corto del cromosoma X (Xp21-22.3) en el que se describe una gran heterogeneidad genética alélica. La patogénesis molecular del síndrome no está bien dilucidada aún.
En Cuba, al no estar disponibles los estudios de genética molecular que permitan identificar las más de 30 mutaciones descritas, el diagnóstico del SNH se realiza mediante la delineación fenotípica del patrón dismórfico, los datos clínicos y las características radiológicas y mediante la utilización del método clínico, tal como se realiza en muchos síndromes de origen genético.2,3 El síndrome SNH aparece en la décima revisión de la Clasificación Estadística Internacional de Enfermedades y Problemas Relacionados con la Salud (CIE-10) con el código Q87.0, en el apartado donde se incluyen los síndromes de malformaciones congénitas que afectan principalmente la apariencia facial. 4
Ulatey otros5) hacen referencia en su artículo a que Walsh y Wegman lo describieron por primera vez en 1937, aunque sin evaluar los aspectos relacionados con la dentición. Posteriormente lo hicieron, en 1974, Horan y Billson6 en Australia y Nance7) y otros en los EE. UU. de forma simultánea. Las anomalías dentarias fueron descritas por primera vez por Seow y otros en 1985.8
En los últimos años se han descrito casos que incluyen en su fenotipo clínico anomalías craneofaciales y alteraciones mentales. La presencia de estructuras dentofaciales atípicas puede ser el primer indicador de otros defectos congénitos presentes en el espectro fenotípico del síndrome, entre las que se incluyen la presencia de dientes supernumerarios, alteraciones en la forma de incisivos y molares, así como signos dismórficos faciales distintivos. Las alteraciones dentales tienen un alto valor diagnóstico y se presentan en un 100 % de los casos, pudiendo afectar tanto la dentición primaria como la permanente.1
En la mayoría de las ocasiones no requieren rehabilitación; sin embargo, se pueden trabajar por razones estéticas, fonéticas o funcionales.9
La odontogénesis es un proceso embrionario mediante el cual las células ectodérmicas del estomodeo se invaginan para formar estructuras, y junto con la interacción del ectomesénquima, forman específicamente los dientes. Se ha descrito la intervención de diversos genes que actúan regulando este proceso. De acuerdo a la fase afectada y genes alterados, se puede dar una perturbación de algunos dientes o de su totalidad, en una sola dentición o en ambas 10)
Las anomalías dentales se clasifican, según su comportamiento morfológico y funcional, en anomalías de número, volumen, posición y forma.11) En la enseñanza de la estomatología cubana también se clasifican de acuerdo al tiempo y la dirección.12
Las anomalías de volumen se refieren a la macrodoncia, según esté aumentado el tamaño mesiodistal de los dientes permanentes o microdoncia según esté disminuido. Las anomalías de posición son aquellas en la que el diente (corona y raíz) se desplaza completamente; son las denominadas gresiones y pueden ser de tipo labial, lingual, mesial, distal, hacia el interior del hueso alveolar o fuera de este.12) Las de forma se originan en la fase de diferenciación morfológica del desarrollo dental; entre las alteraciones que existen debido a estas se encuentran los dientes cónicos o conoides, que consisten en la falta de desarrollo del lóbulo mesio- y distolabial, dando así la apariencia conoide de la corona dental.9
Las de tiempo, por su parte, se refieren a adelantos o retardos en la erupción teniendo en cuenta la edad de brote de dientes temporales y permanentes, mientras que las anomalías de dirección o versión ocurren cuando la raíz queda en posición correcta y lo que se desvía es la corona del diente en sentido horizontal en dirección labial, lingual, mesial o distal. También se incluyen en este grupo las rotaciones que ocurren cuando el diente gira sobre su propio eje vertical.12
El objetivo del presente trabajo fue presentar un caso inusual de anomalías dentarias dentro del espectro fenotípico del síndrome de Nance-Horan. Se solicitó, con estricto cumplimiento de las normas éticas establecidas para ello, el consentimiento informado de la madre para la obtención de las fotos y la publicación del caso.13
Reporte del caso
Paciente masculino de 13 años de edad, de piel blanca, no presentaba trastornos en el aprendizaje. Acudió a la consulta de atención primaria de salud del Policlínico Universitario “Chiqui Gómez Lubían” de la ciudad de Santa Clara, provincia de Villa Clara, Cuba, refiriendo, según sus propias palabras, la presencia de “una cavidad en el canino superior izquierdo temporal en la cual se acumula alimento y percibe fetidez”.
Se recogen antecedentes obstétricos de diabetes gestacional, parto a término, distócico y un peso al nacer de 3128 gramos. Sin presencia de complicaciones perinatales ni historia de consanguinidad parental. Lactancia materna absoluta hasta los siete meses, dieta blanda hasta los dos años de edad, erupción de los dietes temporales normal, actitud pasiva y comportamiento inmaduro.
Antecedentes patológicos personales: catarata congénita y escoliosis dorso-lumbar. Antecedentes familiares: madre que presenta catarata congénita, escoliosis y estrabismo; padre sano.
Presenta como hábitos el cepillado dos veces al día de forma mixta y deglución atípica. Durante el examen facial se definió el tipo facial mediante el uso del cranéometro y se determinó el índice facial, resultando un valor de 108. El resultado anterior permitió concluir que se trataba de un paciente leptoprosopo: una cara larga y estrecha, con forma ovoide y asimetría facial (el tercio inferior se encontraba disminuido con relación al tercio medio). Patrón dismórfico cráneo-facial: frente amplia, nariz grande con un puente nasal alto, perfil facial convexo, orejas grandes (macrotia) con anteversión de la aurícula, piel y mucosas normocoloreadas, articulación témporo-mandibular sin alteraciones. El examen bucal permitió observar en los tejidos blandos: el frenillo labial superior de inserción baja, lengua dentada, los labios con cierre bilabial competente, de color rosa y tamaño normal, surco mentolabial marcado y mentón grande (macrognatia).
Se constató la arcada superior de forma ovoide, al igual que la bóveda palatina, dentición mixta tardía, diastema generalizado, así como un acceso cameral en el canino temporal izquierdo (63). Otros hallazgos fueron: todos los dientes microdónticos, con anomalías de forma: los caninos temporales (53 y 63) con forma de domo o capullo, los premolares presentaban cúspides vestibulares y palatinas separadas por un espacio central amplio, surcos supernumerarios que atravesaban los rebordes marginales, muescas atípicas en las paredes vestibular y palatina.
Como anomalía de tiempo se constató retención de los caninos permanentes, teniendo en cuenta la edad de brote de estos dientes. Como anomalías de dirección: los incisivos centrales y laterales (12, 11, 21, 22) se observaron en vestibuloversión, ligera rotación disto-palatino de 14 y 25, así como rotación disto vestibular de 15 y mesio-vestibular de 24. Se constató una buena higiene dental. (Fig. 1A).
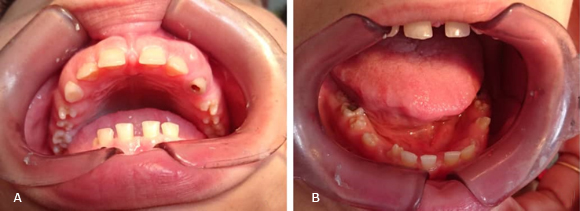
Fig. 1 A. Vista intraoral de la arcada dentaria superior, se observa diastema generalizado, microdoncia dental, anomalías de forma, significativas en los premolares, macrognatismo maxilar y persistencia de los caninos temporales. B. Vista intraoral de la arcada dentaria inferior, se muestra similar a la arcada superior en los primeros tres aspectos, además de la lengua dentada y la erupción de los caninos permanentes.
En la arcada inferior también se observó de forma ovoide, diastema generalizado, obturación oclusal de los primeros molares (36 y 46), dientes microdónticos, linguogresión de los caninos permanentes producto de la persistencia prolongada de los temporales, los premolares presentaban una morfología similar a la de sus homólogos superiores, el segundo premolar inferior derecho (45) poseía una cúspide supernumeraria central que le confería la morfología característica de la malformación dentaria conocida como “diente en mora”, por su parecido con la fruta del mismo nombre. Rotación mesio-lingual del segundo premolar derecho (35) e izquierdo (45) (Fig. 1B).
De acuerdo a la clasificación etiopatológica de las anomalías dentarias, establecida por Mayoral,14 el paciente presenta como anomalía de volumen y forma un macrognatismo maxilar. En oclusión anteroposterior la relación de molares, según la clasificación de Angle, 15 es de distoclusión en la derecha y en la izquierda de neutroclusión, por lo que se clasifica como una Clase II, división I con subdivisión. La clasificación de los caninos no es registrable. El resalte fue de 5 mm, mientras que la curva de Spee fue de 2 mm. En relación transversal las líneas medias coinciden y ambas, a su vez, lo hacen con la línea media de la cara. En el resalte posterior derecho las cúspides palatinas de los dientes posterosuperiores ocluyen en las fosas centrales de los dientes posteroinferiores y las cúspides vestibulares superiores ocluyen por vestibular de las inferiores, en la izquierda tiene una mordida cubierta. En relación vertical el sobrepase es de una corona completa.
Se solicitaron medios auxiliares de diagnóstico como modelos de estudio, fotografías clínicas y radiografías periapicales. En los modelos de estudio se observó un índice incisivo superior de 25,5 y el índice incisivo inferior de 19,9, que en ambos casos expresan microdoncia.
Para calcular la discrepancia hueso-diente se midieron los caninos permanentes directamente en las radiografías. Los resultados fueron: superior 8,1 e inferior 11,8. La anchura transversal, según Mayoral,14 fue: de 14-24: 38,1; de 15-25: 39,2; y de 16-26: 49,5, lo que supone macrognatismo maxilar.
Radiográficamente se observaron los cuatro caninos permanentes retenidos debido a la persistencia de los caninos temporales fuera de la edad normal de exfoliación, dientes con raíces muy cortas y cámaras pulpares amplias (Fig. 2). El plan de tratamiento consistió en mioterapia, exodoncia de los cuatro caninos temporales e interconsulta con el especialista en genética clínica.
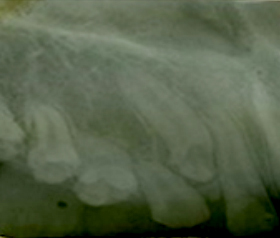
Fig. 2 Radiografía periapical. Se constatan raíces cortas y cámaras pulpares amplias, el canino permanente superior derecho impactado producto de la persistencia del canino temporal.
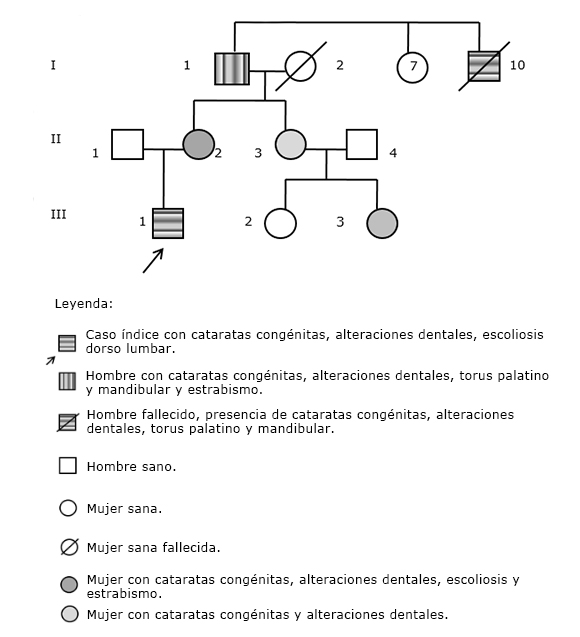
Con la información brindada por la madre se confeccionó el árbol genealógico hasta tres generaciones partiendo del paciente como propositus o caso índice (III.1) (Fig. 3).
Discusión
De acuerdo a los últimos datos estadísticos publicados por la Organización Nacional de Enfermedades Raras (NORD, del inglés: National Organization for Rare Disorders) en la literatura médica internacional existen menos de 50 familias en la que se ha descrito el SNH a nivel mundial.16
Las mutaciones en el gen SNH tienen un efecto pleiotrópico, por cuanto existe una alta expresión de su producto proteico en el cerebro medio, retina, cristalino y dientes, lo que resulta en un patrón dismórfico dado por cara alargada, nariz grande con un puente nasal alto, prognatismo mandibular y orejas grandes (macrotia) y displásticas con anteversión de la pinna. Las anomalías oculares incluyen catarata congénita bilateral, presente en un 100 % de los casos, microcórnea (96 %) y microftalmia. En aproximadamente el 93 % de los casos las anomalías oculares causan un severo nistagmo y, algunas veces, estrabismo (43 %). Un 89 % de los afectados requiere de procedimientos quirúrgicos.2
Los molares son redondeados, globulares y se ven como flores, con presencia de una cúspide central supernumeraria que da forma de mora a las piezas afectadas. Los incisivos se describen con una muesca típica en el centro del borde incisal, denominados “dientes de Hutchinson” y algunos autores lo nombran también “incisivos en forma de destornillador”.17 En el 65 % de los casos aparecen dientes supernumerarios.2,5
Características como la retención prolongada de los dientes deciduos, anomalías pulpares como taurodontismo, cámaras pulpares amplias y cálculos pulpares, son encontrados comúnmente en el SNH.2) La presencia de diastemas es un rasgo característico reportado en todos los casos que se han descrito.5
Los antecedentes patológicos personales del paciente, el patrón dismórfico cráneo facial y las radiografías coinciden con características de otros casos de SNH reportados en la literatura.2,5,17,18,19
La mutación del gen SNH se expresa completamente solo en los varones, y, de ellos, entre el 20-30 % presentan diferentes grados de discapacidad intelectual. Otros hallazgos ocasionales incluyen los síntomas del espectro autista, retraso del desarrollo psicomotor y otras alteraciones neuropsicológicas como los trastornos de conducta (agresión, estereotipias), además de braquidactilia, braquimetacarpia, entre otros.17
En el árbol genealógico del paciente estudiado se puede apreciar una gran expresividad variable, así, en el abuelo enfermo del propositus (I.1) además de cataratas congénitas y anomalías dentales, se constató la presencia de torus palatino y mandibular y estrabismo; mientras que en el tío abuelo fallecido (I.10) no se refirió la existencia de estrabismo. Como los varones son hemicigóticos para los genes ligados al cromosoma X, basta con una copia del alelo mutado para que aparezca una enfermedad de herencia recesiva ligada al sexo, mientras que las mujeres, al poseer dos copias del cromosoma X, requieren la presencia de dos alelos mutados (homocigóticas), lo que explica la mayor frecuencia de varones afectados por trastornos recesivos ligados al cromosoma X.
Sin embargo, en la familia de este paciente se identificaron mujeres (madre, tía materna y prima hermana) con grados variables de severidad clínica, tal como se muestra en la genealogía (II.2, II.3 y III.3, respectivamente). Esta situación que también es descrita por otros investigadores,5,17,18 lo que puede ser explicado por el fenómeno de lyonización desfavorable: al inactivarse en las células somáticas de estas mujeres una mayor proporción de cromosomas X que presentan el alelo normal o salvaje del gen SNH y quedan activos aquellos que portan el alelo mutado. Por lo insuficientemente estudiadas que resultan las bases moleculares de este síndrome aún, otra posible explicación estaría en el hecho de una trasmisión hereditaria dominante ligada al cromosoma X.
En la descendencia de los hombres afectados todas las hijas serán heterocigotas, pues le transmitirán el cromosoma X con el alelo mutado, mientras que ninguno de los hijos varones estaría afectado, ya que a ellos les transmitirían el cromosoma Y. Por su parte, las mujeres heterocigotas (portadoras sanas) tienen un 50 % de posibilidades de transmitir el gen mutado: un 25 % de tener hijos varones afectados y un 25 % de tener hijas portadoras sanas.19
En apoyo a la evolución del paciente, por la parte estomatológica, se le instruyó a él y a sus familiares en medidas de autocuidado y se le explicó la importancia de realizar visitas periódicas al estomatólogo y se hizo énfasis en el control de su higiene bucal. En el paciente se tiene en cuenta el uso de sellantes de fosas y fisuras, de ser necesarios, así como la utilización de lacas y barnices para proteger su esmalte. Por la parte médica se orientaron interconsultas. Al momento de la escritura de este artículo se mantiene en seguimiento por las siguientes especialidades: ortopedia, oftalmología y genética clínica.
Entre las fortalezas asociadas a este reporte se incluyen la adecuada interdisciplinaridad existente entre las diferentes especialidades médicas y estomatológicas que permitieron el manejo integral y el adecuado proceso de asesoramiento genético a la familia. Entre las limitaciones se encuentran la imposibilidad de realizar en nuestro medio estudios genéticos que permitan la confirmación del diagnóstico molecular de este síndrome y, por ende, la imposibilidad de ofrecer un diagnóstico prenatal por métodos directos o indirectos en próximos embarazos de la madre del paciente.
Conclusiones
Las anomalías dentales pueden presentarse como una característica asociada a un síndrome específico, en este caso, el síndrome Nance-Horan, por lo cual es de crucial importancia realizar un cuidadoso examen, tanto clínico como radiográfico de los pacientes con anomalías como las descritas en la literatura. Al reconocer estas enfermedades se puede ofrecer atención interdisciplinaria sobre la base de los hallazgos y establecer el patrón de herencia para brindar asesoramiento genético familiar oportuno. Se insiste en el trabajo mancomunado entre las diferentes disciplinas y especialidades, tanto médicas como estomatológicas.

