










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Human papillomaviruses (HPV) are the etiological agents of cervical and other anogenital malignancies.(1) HPV is a small double-stranded DNA virus with a circular genome of approximately 8 kb. The viral icosahedral capsid is formed by 72 pentameric capsomeres, which consist of L1 and L2 molecules. The L1 is the major capsid protein with molecular weight of 55 kDa.2,3 Recombinant production of L1 using heterologous systems has resulted in the formation of virus-like particles (VLPs) that resemble the native virions in size and shape but lack the potentially oncogenic viral genome.4,5 Assembled VLPs are potent immunogens, as they can induce high titers of neutralizing antibodies for preventing HPV infections.4,6
Currently there are three licensed preventive HPV vaccines available on the market: Gardasil, Gardasil-9, and Cervarix. The three HPV vaccines target HPV type 16 (HPV-16) and HPV type 18 (HPV-18),7 which are responsible for 69.4 % of cervical cancer cases.8 HPV-16 is the most common viral genotype detected in cervical cancers; however, the frequencies of HPV-18 in adenocarcinomas and adenosquamous carcinoma are known to be higher than those of HPV-16.9,10 Thus, HPV-18 is a representative of the high-risk viruses most often associated with genital carcinoma.11 The HPV vaccines are based on L1 VLPs, obtained in eukaryotic hosts with high production costs, which have affected their availability in low-income countries, where cervical cancer results in high mortality. The quest for a cheaper, thermostable and broad spectrum vaccine has led to the development of new prophylactic vaccine candidates obtained from more economical alternative expression hosts such as Escherichia coli and methylotrophic yeasts.12,13
Escherichia coli has been the most widely used bacterial system for the recombinant production and characterization of HPV L1 protein.14,15 Most of the studies of the HPV L1 gene expression in E. coli deal with the generation of HPV-16 and HPV-11 L1 proteins fused to N-terminal glutathione S-transferase (GST), which promotes correct folding and stability of recombinant L1 in E. coli.15,16,17 However, among the HPV L1 proteins targeted by the commercial vaccines, HPV-18 L1 has the highest amount of amino acids residues and cysteine residues along its sequence18,19 and its expression in E. coli has been less studied than the HPV-16 or HPV-11 L1 genes. Seo et al. obtained VLP from the purified full-length HPV-18 L1, which was expressed as a GST-18 L1 fusion,20 while Gu et al. reported the production of the HPV-18 L1, truncated at the amino terminus, in the soluble fraction of E. coli. In this last work, a bivalent HPV vaccine candidate against genotypes 16 and 18 has been developed,13 which is currently undergoing a Phase 3 clinical study (ClinicalTrials.gov NCT01735006) and supports the potentiality of E. coli as a host for obtaining VLPs of HPV. In addition, previous results have shown a positive effect of the carboxy terminal truncation of the HPV-16 L1 protein on its production and solubility in E. coli and insect cells. Schädlich et al obtained an almost twofold increase in HPV-16 L1 protein yields in constructs with C-terminal deletions in E. coli,21 while Deschuyteneer et al reported that removal of the carboxy terminus of the HPV-16/18 L1 proteins might minimize the risk of cellular nucleic acid encapsidation for obtaining the truncated proteins in insect cells as active ingredients of Cervarix®.22
In Cuba, several molecular epidemiologic studies of HPV infections have found the distribution of oncogenic HPV genotypes in women,23,24,25,26,27 in patients with colorectal cancer28 and more recently, in HIV-seropositive men.29 In women, the most recent study detected 30 different HPV genotypes in 519 participants aged between 15 and 59 years, where the most prevalent type was HPV-16 (41.0 %), followed by HPV-31 (11.6 %) and HPV-18 (10.2 %).27 In patients with colorectal tumors, the infection frequency was 25 % and genotypes 16 and 33 were identified. Viral infection was associated with the presence of adenocarcinomas (41.7 %, p = 0.009)28 In anogenital samples from 56 HIV-infected men, a high frequency of HPV infection, above 50 %, was detected. HPV- 16 genotype was the most common type (52 %), while HPV-18 was the most frequently detected genotype in men with high-grade squamous intraepithelial lesions.29 Therefore, considering that HPV genotype 18 is a high-risk type with clinical relevance in Cuba, the aim of this work was to clone a HPV-18 L1 gene from a Cuban female patient and to express the full-length and deletion variants of the HPV-18 L1 gene in E. coli, for further vaccine candidate development.
METHODS
Clinical sample and total DNA extraction
To isolate the HPV-18 L1 gene, total DNA was purified from HPV-18-infected cervical cells, which were collected by cervical scrapings from a 59 year-old woman suffering from epidermoid carcinoma (La Habana, Cuba). The cell sample had been previously diagnosed as HPV-18 positive by the CLART® HPV2 assay (Genómica S.A.U., Spain).30 Total DNA was isolated by the GFX Genomic Blood DNA Purification Kit (GE Healthcare, USA) using the Direct Method, according to the manufacturer´s specifications.
Amplification and cloning of a full-length HPV 18 L1 gene
Total DNA was used to amplify by PCR a 1521 bp DNA fragment, comprising the full-length HPV-18 L1 gene, with the forward primer 5′-CATATGGCTTTGTGGCGGC-3′ (18L1F), including the NdeI restriction enzyme site, and the reverse primer 5′-AGATCTATTACTTCCTGGCACGTACAC -3′ (18L1R), containing the BglII site at the 3′ terminal sequence (underlined sequences are the restriction enzyme sites). Both sites were included to enable the subsequent cloning of the L1 amplified product. The NdeI restriction site was incorporated in the forward primer for introducing a translational start codon, coding for a consensus methionine required for efficient formation of VLP in recombinant expression systems.19 The primers were designed from the alignment of HPV-18 L1 gene sequences of the 10 HPV-18 representative genomes for viral variant sublineages.31
The PCR conditions included 35 cycles of 30 sec at 94 °C, 30 sec at 60 °C and 3.5 min at 72 °C on a Master Cycler (Eppendorf, Germany). The amplified product was purified with the Wizard® SV Gel and PCR Clean-Up System (Promega, USA) and was cloned into the pGEM-T Easy vector (Promega, USA) in E. coli Mach 1-T1, obtaining pGEM-T Easy-HPV-18L1 plasmid. The recombinant plasmid was sequenced using the Macrogen services (Korea) to determine the cloned HPV-18 L1 nucleotide sequence. The identity of the nucleotide sequence was established using the Blastn bioinformatic program (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome).
Construction of expression plasmids encoding the full-length and truncated variants of HPV-18 L1 proteins in E. coli
To evaluate the expression of the full-length HPV 18 L1 gene in E. coli, the 1530 bp NdeI - BglII fragment was excised from pGEM-T Easy-HPV18L1 plasmid and subcloned into pET26b vector to generate pET26b-HPV-18 L1.
For evaluating the effect of N- and C-terminal truncations on the production and solubility of HPV-18 L1 protein, three deletion mutants were constructed by PCR using pET26b-HPV-18 L1 as template. A genetic construct encoding a carboxy-terminal-only truncated variant (L1(C30 , missing aa 478-507) was obtained, which was generated by PCR amplification with the primer 18L1F and the reverse primer ∆30 (5´- AGATCTATTAGCGACGCAATCCAGCCTG-3´). The last primer hybridizes to the region previous to the last 30 codons of the HPV 18 L1 cloned gene.
N-terminal truncation of the HPV-18 L1 protein was also evaluated. To select the number of amino acids to be removed at the amino terminus of the HPV-18 L1 protein, we estimated the translation initiation rate (TIR) of the HPV-18 L1 gene, with 2, 3, 4, 5, 6, 7, 8 and 9 codons deleted at its 5´end, by using the RBS Calculator (https://salislab.net/software/)32 and the UTR Designer (http://sbi.postech.ac.kr/utr_designer)33 tools, in reverse-engineering mode. Based on the predictions of these tools, two other truncation mutants of HPV-18 L1 protein were generated. Thus, deletions of the coding region for the first 5 ((N5L1, aa 6-477) and 6 ((N6L1, aa 7-477) amino acids, were combined with a C-terminal 30 amino acids deletion. PCR amplifications were carried out with oligonucleotide (30, in combination with primers 5´-CATATGCCATCAGACAATACCGTATACCTTCC-3´ or 5´- CATATGTCAGACAATACCGTATACCTTCCACCTC-3´, for obtaining (N5L1(C30 and (N6L1(C30, respectively. NdeI and BglII recognition sites (underlined) were added to the primers to facilitate subcloning. The three PCR fragments were independently ligated into the pGEM-T Easy vector (Promega, USA) and their nucleotide sequences were verified. Then, the three NdeI/BglII fragments, containing the truncated sequences L1(C30, (N5L1(C30 and (N6L1(C30 of the HPV18 L1 gene from the pGEM-T Easy derivatives, were independently ligated into NdeI/BglII digested pET26b vector to generate pETHPV18L1(C30, pETHPV18(N5L1(C30 and pETHPV18(N6L1(C30, respectively. E. coli Mach 1-T1 was used for the cloning steps, while E. coli BL21(DE3) and E. coli SHuffle T7 were used as expression hosts. Ampicillin (100 μg/mL) and kanamycin (100 μg/mL) were used when necessary.
Plasmid pET28a-HPV-18L1-tag, encoding the HPV-18 L1 protein fused to the histidine tag and N terminal amino acids derived from the vector (Histag-HPV-18L1),34 was also introduced in E. coli BL21 (DE3) and E. coli SHuffle T7 in order to compare the production of the Histag-HPV-18L1 and the three L1 truncated mutants, obtained above.
Evaluation of HPV-18 L1 protein production in E. coli BL21 (DE3) and E. coli SHuffle T7
Production of the HPV18 L1 protein variants was promoted by conventional induction with isopropyl-h-D-thiogalactopyranoside (IPTG). E. coli BL21 (DE3) and E. coli SHuffle T7 clones, harboring pET26b-HPV-18 L1, pETHPV18L1(C30, pETHPV18(N5L1(C30, pETHPV18(N6L1(C30 or pET28a-HPV-18L1-tag34 were grown overnight in terrific broth medium (TB) (35). Fifty microliters of pre-cultures were inoculated into 5 mL of TB medium and cells were incubated at 28 °C and 220 r.p.m. until reaching 0.6-0.8 units of optical density (at 600 nm, OD600), moment in which IPTG (0.01; 0.1 or 1 mM as final concentration) was added to the culture broth for inducing recombinant protein expression. Then, cultures were incubated at 28 °C and 220 r.p.m. for 4 h and samples were withdrawn every 2 h. Lastly, cells were harvested and stored at -20 °C until further use.
For evaluating L1 protein production under auto-induction conditions, cells were cultured overnight under non-inducing conditions into modified TB medium.36 Production cultures, composed by the abovementioned TB medium supplemented with 2 g/L lactose or IPTG (125, 60 or 30 µM), were inoculated at an initial OD of 0.1 and allowed to grow for 14 h at 28 °C and 220 r.p.m. Cell samples were processed as mentioned before.
Evaluation of HPV18-L1 protein solubility in E. coli BL21 (DE3) and E. coli SHuffle T7
Samples were incubated 15 min at room temperature in a lysozyme solution at 20 mg/ml. Partially broken cells were freezed-thawed 3 times to promote cell lysis. After a centrifugation step at 12,000 × g for 20 min, the soluble fractions were frozen and the insoluble fractions were washed and resuspended in equivalent amounts to the soluble fractions. All protein fractions were conserved at -20 (C for further SDS-PAGE and Western blot analysis.
SDS-PAGE and Western blot analysis
For estimation of molecular weight and determination of identity and solubility of HPV18 L1 proteins, 10 % (w/v) sodium dodecyl sulphate polyacrylamide gel electrophoresis (10 % SDS-PAGE) under reduced conditions,37 followed by staining with Coomassie brilliant blue ,was carried out. Cell samples were homogenized to 6.5 units of OD600 with phosphate-buffered saline.38 For Western blot analysis, the proteins resolved on the gels were transferred onto protran nitrocellulose membranes (Schleicher and Schuell Bioscience, Germany). The membranes were blocked with PBST solution38 containing 5 % w/v nonfat dry milk and then reacted with mouse anti-HPV16 L1 antibody (clone CamVir-1, Merck, USA) diluted 1:15 000. Then, membranes were incubated with anti-mouse IgG-HRP (1:64 000, CIGB SS, Cuba). Immunoreactive protein bands were visualized using peroxidase substrate 3,3'-Diaminobenzidine (DAB, Sigma, USA). The densitometric analysis of protein bands on SDS-PAGE gels and Western blots scans were done with Gel Analyzer 2010 software.39 All data represents mean values of at least three independent experiments.
Detection of inclusion bodies formation in E. coli SHuffle T7 (pET28a-HPV-18L1-tag( by transmission electron microscopy (TEM)
The cells expressing the full-length HPV-18 L1 gene fused to a His-Tag coding sequence were cultivated at 28 °C for 4 h upon 0.1 mM IPTG induction. Then the cells were harvested, fixed with 2 % glutaraldehyde and 1 % osmium tetroxide, both in 0,1 M sodium phosphate buffer solution at pH 7,2. After graded-acetone serial dehydration steps, the cells were embedded in Spurr resin, ultrathin sectioned, stained with uranyl acetate and lead citrate solutions, and then observed with a Jeol JEM 1011 (JEOL, Japan) transmission electron microscope at 20 000 X.
RESULTS
Amplification and cloning of the full-length HPV 18 L1 gene in Escherichia coli
PCR amplification using total DNA isolated from a cervical exfoliated cell sample and the primer pairs 18L1F and 18L1R (Fig. 1, A) produced an amplicon of ~1.5 kb (Fig. 1, B, lane 1), size close to the expected value (1524 bp, Fig. 1, A). The identity of the PCR fragment was analysed by digestion with the restriction enzymes KpnI, BamHI and XbaI, which recognize conserved and unique sequences in the HPV18 L1 reference sequence (accession number NC(001357, Fig. 1, A). As expected, the Kpn I enzyme produced two bands of ~650 and ~800 bp (Fig. 1, B, lane 2), values close to the expected values (662 bp and 862 pb; Fig. 1, A). Digestion of the amplified fragment with BamHI enzyme also produced two bands (~200 bp and ~1300 pb; Fig. 1, B, line 3), close to the predicted values of 206 pb and 1318 bp (Fig. 1, A). Digestion with XbaI generated two bands of ~1 300 pb and ( 150 bp (Fig. 1, B, line 4), close to the predicted sizes of 1 405 pb and 119 bp (Fig. 1, A), respectively. These results showed the conservation of three different restriction enzyme recognition sites in the amplified fragment and revealed the similarity with the reference HPV18 L1 gene at the nucleotide level. Then, the PCR product was cloned into the pGEM-T Easy vector, obtaining the plasmid pGEM-T Easy-HPV18L1, which was sequenced. The DNA sequence analysis revealed that the cloned HPV-18 L1 gene was 99.9 % similar to the African variant EF202152, representative for the HPV-18 sublineage B3. There was only one nucleotide difference between the EF202152 (A) and the cloned sequence (G) at position 1518, but without impact on the translated amino acid sequence. The complete nucleotide sequence of the cloned HPV-18 L1 gene was deposited in GenBank under the accession number MK211166.
 A: Schematic representation of HPV18 L1 reference sequence (accession number NC(001357) with the unique restriction sites Xba I, Kpn I and BamHI included. The primers 18L1F and 18L1R were also represented. B: Restriction analysis of the generated amplicon lane M: DNA molecular weight marker (SERVA DNA Standard 100 bp DNA Ladder extended); lane 1, PCR product; lanes 2-4, PCR product digested with KpnI, BamHI and XbaI, respectively.
A: Schematic representation of HPV18 L1 reference sequence (accession number NC(001357) with the unique restriction sites Xba I, Kpn I and BamHI included. The primers 18L1F and 18L1R were also represented. B: Restriction analysis of the generated amplicon lane M: DNA molecular weight marker (SERVA DNA Standard 100 bp DNA Ladder extended); lane 1, PCR product; lanes 2-4, PCR product digested with KpnI, BamHI and XbaI, respectively.Fig. 1 Amplification of the full-length HPV-18 L1 gene using total DNA isolated from a cervical exfoliated cell sample.
Subcloning of the full-length HPV18 L1 gene and expression in E. coli BL21(DE3)
The cloned HPV-18 L1 gene was subcloned into pET26b vector and the recombinant plasmid pET26b-HPV-18 L1 was introduced initially into E. coli BL21(DE3), considering that this strain lacks OmpT outer membrane protease, which has been reported to degrade the carboxy terminus of HPV-16 L1 protein.40 Expression of the HPV18L1 gene in response to increasing amounts of IPTG (0.01; 0.1 and 1.0 mM) in TB medium was analyzed by SDS-PAGE. Under these conditions, SDS-PAGE analysis did not show overexpression of HPV-18 L1 protein in IPTG-induced E. coli BL21(DE3) (pET26b-HPV-18 L1(, since there were not differences observed between induced and non-induced E. coli cells (Fig. 2, A, lanes 1 vs. 2-7). However, an HPV-18 L1-specific immunoreactive band of about 57 kDa could be detected in whole cell lysates of the recombinant strain when IPTG was added to a final concentration of 1 or 0.1 mM, both for 2 and 4 h of induction (Fig. 2, B, lanes 4-7). Immunoreactive bands of lower molecular weights were also detected, which may be due to proteolytic degradation or early translation termination, as it has been previously reported for HPV16 L1.16,40 HPV-18 L1 was faintly detected in whole cell lysates of E. coli BL21(DE3) [pET26b-HPV-18 L1] at the lowest inducer concentration used (0.01 mM) (Fig. 2, B, lanes 2 and 3). As expected, there was no detectable HPV-18 L1-specific immunoreactive bands in cell lysate of the non-induced control (Fig. 2, B, lane 1). The results revealed that E. coli BL21(DE3) was able to produce the native HPV18-L1 protein, although at low levels.
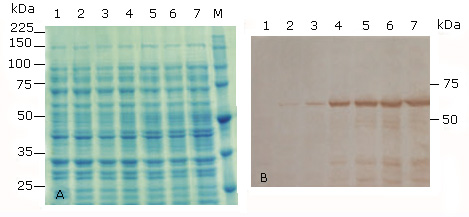 A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, without IPTG (un-induced control); lane 2, 0.01 mM IPTG 2 h; lane 3, 0.01 mM IPTG 4h; lane 4, 0.1 mM IPTG 2 h; lane 5, 0.1 mM IPTG 4 h; lane 6, 1 mM IPTG 2h; lane 7, 1 mM IPTG 4 h; lane M, Broad-range protein molecular weight markers (Promega, USA).
A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, without IPTG (un-induced control); lane 2, 0.01 mM IPTG 2 h; lane 3, 0.01 mM IPTG 4h; lane 4, 0.1 mM IPTG 2 h; lane 5, 0.1 mM IPTG 4 h; lane 6, 1 mM IPTG 2h; lane 7, 1 mM IPTG 4 h; lane M, Broad-range protein molecular weight markers (Promega, USA).Fig 2 Production of the full-length HPV-18 L1 at different time of induction and concentrations of IPTG in E. coli BL21(DE3) [pET26b-HPV-18 L1].
Production of truncated variants of HPV-18 L1 in E. coli BL21(DE3)
We next evaluated the contribution of the deletion of the amino terminus of the native HPV-18 L1 protein on its production and solubility. For selecting the number of amino acid to be remove from the amino-terminus, translation initiation rates of L1 genes with deletions from one to nine codons was estimated by using the RBS Calculator and the UTR Designer. According to predictions of the two programs, the L1 truncated variants with higher TIR´s values were those coding for the proteins lacking the first five and six amino acids ((N5L1 and (N6L1; table). Both DNA constructs had TIR higher than the values of the full-length HPV-18 L1 gene (table). Therefore, we decided to amplify by PCR the HPV-18 L1 gene with deletions of five and six codons at the 5´ end of the cloned L1 gene. For both mutants, deletions of 30 codons at the 3´ end of the wild-type HPV-18 L1 gene were generated. We also evaluated a construction with the last 30 codons deleted and the wild-type amino terminus (L1(C30) to assess the contribution of the deletion of the carboxy terminus of the native L1 protein on its production and solubility.
We successfully amplified the PCR fragments corresponding to the three deletion variants of HPV-18 L1 gene, cloned them into the pGEM-T Easy Vector (Promega, EUA) and subcloned them into the expression vector pET26b. The resulting recombinant plasmids pETHPV18(N5L1(C30, pETHPV18(N6L1(C30 and pETHPV18L1(C30 were independently introduced in E. coli BL21(DE3). We also evaluated E. coli BL21(DE3) (pET28a-HPV-18L1-tag(, a His-Tagged fusion derivative of the cloned HPV-18 L1 gene, which produced L1 protein (Histag-HPV-18L1) at detectable levels in SDS-PAGE,34 in agreement with the higher TIRs values estimated by the two calculator, respect to the full-length L1 gene (table).
Table Translation initiation rates (TIR) of constructs coding for N-terminal truncated and 6xHis-tagged (Histag-L1) variants of HPV-18 L1 protein, estimated by the RBS Calculator and the UTR Designer tools

The ribosome binding site calculators were used in the reverse engineering mode and in agreement with the requirements of each tool.
The expression of the truncated- and Histag- HPV18 L1 variants was induced in the same medium and culture conditions than for E. coli BL21(DE3) [pET26b-HPV-18 L1], but IPTG was used at 0.1 mM. According to SDS-PAGE results, the three truncated HPV-18 L1 variants were produced as faint bands in E. coli BL21(DE3) and at similar levels (Fig. 3, A, lanes 2-4 & 7-9), which was confirmed by densitometric analysis of the immunoblots (Fig. 3, B, lanes 2-4 & 7-9). The fact that the carboxy-terminal-only truncated variant, L1(C30, was produced in E. coli BL21(DE3) at similar levels than the HPV-18 L1 protein truncated at both termini suggested that the carboxyl terminus truncation was the deletion that contributed to increase the HPV-18 L1 protein amounts. The densitometric analysis of blots also showed that the production of the three truncated forms of HPV-18 L1 had a fourfold improvement in L1 levels, compared to the wild-type HPV-18 L1 protein (Fig. 3, B, lanes 1-4 & 6-9). From a solubility point of view, the three truncated variants of HPV-18 L1 had similar solubility, with at least a thirty-five percent of the L1 in the soluble fraction, compared to the wild-type HPV-18 L1 protein; which was mainly found in the insoluble fraction (92 % ± 0.8) (Fig. 3, B, lane 6). In accordance with the predictived TIR results obtained by the RBS Calculator, the higher amount of HPV-18 L1 was obtained when the wild-type HPV-18 L1 gene was fused to the 5´ end of the His-Tag coding sequence, carried by pET28a (Fig. 3, B, lanes 5 and 10). According to densitometric studies on Western blot, the production of Histag-HPV-18L1 resulted in about a tenfold increase in protein amount compared to the wild-type HPV-18 L1; although, the fusion protein was mostly found in the insoluble fraction (99 % ± 0.4).
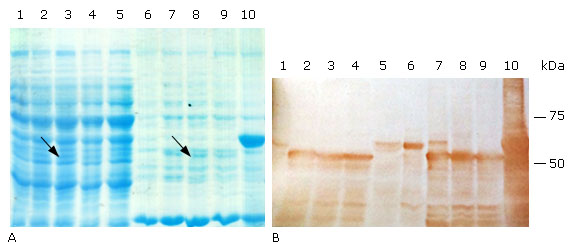 A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, pET26b-HPV-18 L1 soluble fraction (sf); lane 2, pETHPV18L1(C30 sf; lane 3, pETHPV18(N6L1(C30 sf; lane 4, pETHPV18(N5L1(C30 sf; lane 5, pET28a-HPV-18L1-tag sf; lane 6, pET26b-HPV-18 L1 insoluble fraction (if); lane 7, pETHPV18L1(C30 if; lane 8, pETHPV18(N6L1(C30 if; lane 9, pETHPV18(N5L1(C30 if; lane 10, pET28a-HPV-18L1-tag if. The arrows in lanes 3 and 8 (3A) show the bands corresponding to an expected truncated variant ((N6L1(C30).
A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, pET26b-HPV-18 L1 soluble fraction (sf); lane 2, pETHPV18L1(C30 sf; lane 3, pETHPV18(N6L1(C30 sf; lane 4, pETHPV18(N5L1(C30 sf; lane 5, pET28a-HPV-18L1-tag sf; lane 6, pET26b-HPV-18 L1 insoluble fraction (if); lane 7, pETHPV18L1(C30 if; lane 8, pETHPV18(N6L1(C30 if; lane 9, pETHPV18(N5L1(C30 if; lane 10, pET28a-HPV-18L1-tag if. The arrows in lanes 3 and 8 (3A) show the bands corresponding to an expected truncated variant ((N6L1(C30).Fig. 3 Soluble production of truncated and 6xHis-tagged variants of HPV-18 L1 in E. coli BL21(DE3).
Production of truncated and 6xHis-tagged variants of HPV-18 L1 in E. coli SHuffle T7
Considering that the three truncated HPV-18 L1 proteins were partially found in the soluble fraction of E. coli BL21(DE3), and HPV-18 L1 and its derived proteins contain 14 cysteine residues potentially involved in the formation of disulfide bridges,18E. coli Shuffle T7 was selected as host to evaluate the production of L1 proteins in the soluble fraction. Therefore, plasmids pETHPV18(N5L1(C30, pETHPV18(N6L1(C30 and pETHPV18L1(C30, as well pET28a-HPV-18 L1-tag were transformed in E. coli SHuffle T7. The induction of protein expression was carried out in the same medium and culture conditions as for E. coli BL21(DE3).
In Coomassie stained polyacrilamide gels, the the three truncated proteins were not visually detected in cellular fractions of recombinant E. coli SHuffle T7 (Fig. 4, A, lanes 2-4 & 7-9), but the host protein background was high around the molecular weight of the truncated proteins in the soluble fractions (Fig. 4, A, lanes 1 & 5 vs. 2-4). This fact, combined with the finding that Histag-HPV-18L1 was detected as a faint band in the insoluble fraction of E. coli SHuffle T7 (Fig. 4, A, line 10) and as overexpressed band in E. coli BL21(DE3) (pET28a-HPV-18L1-tag( (Fig. 3, A, line 10), suggested that the truncated proteins were produced in smaller amounts in E. coli SHuffle T7 than in E. coli BL21(DE3), where the truncated proteins were detected by Coomassie staining (Fig. 3, A, lines 2-4). According to densitometric analysis on Western blot, the amounts of the L1(C30, (N5L1(C30 and (N6L1(C30 produced by E. coli SHuffle T7 were at least two times higher than that of the native protein HPV-18 L1, encoded by pET26b-HPV-18 L1. L1(C30 was produced at similar levels than (N5L1(C30 and (N6L1(C30 (Fig. 4, B, lanes 2-4 & 7-9), as it was observed in E. coli BL21(DE3), and confirmed that the carboxy-terminal truncation of the HPV-18 L1 was the deletion that led to an increase in the HPV-18 L1 amount in E. coli. The truncated proteins L1(C30, (N5L1(C30 and (N6L1(C30 had similar solubility profiles and they were detected in a percentage higher than 50 % in the soluble fraction of E. coli SHuffle cytoplasm (Fig. 4, B, lanes 2-4 vs. 7-9), which was higher than the proportion obtained in E. coli BL21(DE3). Additionally, a clear band of about 50 kDa was detected at lower levels than that the truncated full size proteins in the soluble fractions, which were not present in the insoluble fractions of E. coli SHuffle T7 (Fig. 4, B, lanes 2-4 vs. 7-9) nor in the cellular fractions of E. coli BL21(DE3). This phenomenon could be related to the proteolytic activity of some proteases like OmpT, which is present in the genome of E. coli SHuffle T7, a K-12 E. coli derivative.40,41
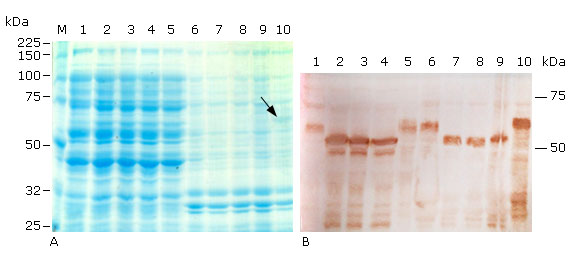 A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, pET26b-HPV-18 L1 soluble fraction (sf); lane 2, pETHPV18L1(C30 sf; lane 3, pETHPV18(N5L1(C30 sf; lane 4, pETHPV18(N6L1(C30 sf; lane 5, pET28a-HPV-18L1-tag sf; lane 6, pET26b-HPV-18 L1 insoluble fraction (if); lane 7, pETHPV18L1(C30 if; lane 8, pETHPV18(N5L1(C30 if; lane 9, pETHPV18(N6L1(C30 if; lane 10, pET28a-HPV-18L1-tag if; M, Broad-range protein molecular weight markers (Promega, USA). The arrow in lane 10 (4A) shows the band corresponding to the expected Histag-HPV-18L1.
A: 10 % SDS-PAGE stained with Coomassie blue R-250, and B: Immunoblotting analysis using the HPV-16 L1 antibody CAMVIR-1. Lane 1, pET26b-HPV-18 L1 soluble fraction (sf); lane 2, pETHPV18L1(C30 sf; lane 3, pETHPV18(N5L1(C30 sf; lane 4, pETHPV18(N6L1(C30 sf; lane 5, pET28a-HPV-18L1-tag sf; lane 6, pET26b-HPV-18 L1 insoluble fraction (if); lane 7, pETHPV18L1(C30 if; lane 8, pETHPV18(N5L1(C30 if; lane 9, pETHPV18(N6L1(C30 if; lane 10, pET28a-HPV-18L1-tag if; M, Broad-range protein molecular weight markers (Promega, USA). The arrow in lane 10 (4A) shows the band corresponding to the expected Histag-HPV-18L1.Fig. 4 Soluble production of truncated and 6xHis-tagged variants of HPV-18 L1 achieved under conventional IPTG induction in E. coli SHuffle T7.
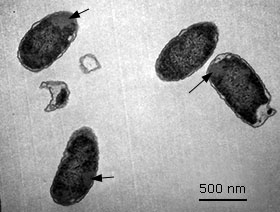
Histag-HPV-18L1 protein was mainly detected in the E. coli SHuffle T7 insoluble fraction (Fig. 4, A and 4, B, lanes 5 and 10), which suggested that the fusion protein was also forming insoluble aggregates or it was deposited in inclusion bodies (Ib), similar to the full-length HPV-18 L1 (Fig. 4, A and 4, B, lane 1 and 6). Taking into account that the Histag-HPV-18L1 protein was obtained at higher amounts than the full-length HPV-18 L1 in the insoluble fraction (Fig. 4, B, lanes 6 vs. 10), the E. coli SHuffle T7 [pET28a-HPV-18L1-tag] cells were analyzed by TEM. This analysis revealed the presence of Ib (Fig. 5), which indicated that the Histag-HPV-18L1 protein was aggregated in Ib in E. coli SHuffle T7.
Production of the HPV-18 L1 variant truncated only at the carboxy terminus by E. coli SHuffle T7 in auto-induction conditions
Considering the low levels of HPV-18 L1 produced by E. coli SHuffle T7 when it was cultured under conventional induction with IPTG, we evaluated its production under auto-induction conditions, with lactose or IPTG as autoinducers.36,42 The variant truncated only at the carboxy terminus L1(C30 was selected for this study, since it carries the deletion that contributed to increase the amounts and solubility of this recombinant protein under conventional IPTG induction (Fig. 4).
At 14 h of auto-induction, immunoreactive bands with the expected size (~52 kDa) were detected in whole cell lysates of the recombinant strain (Fig. 6, A, lanes 4 and 6-8). The relative band intensities of L1(C30 were similar at all tested IPTG concentrations in auto-induction conditions (125 µM, 60 µM and 30 µM) (Fig. 6, A, lanes 6-8). These results suggested that there is no a linear relationship between the IPTG concentration in autoinduction conditions and the expression levels of the truncated HPV-18 L1 in E. coli SHuffle T7, which was in agreement with the fact that the E. coli Shuffle T7 is not a lacY mutant. According to densitometric analysis, E. coli SHuffle T7 produced approximately three times more L1(C30 under auto-induction with lactose, than by conventional induction with IPTG (Fig. 6, A, lanes 2 vs. 4). Thus, E. coli SHuffle T7 produced levels of L1(C30 comparable to those obtained under conventional induction with IPTG by E. coli BL21(DE3). The solubility of L1(C30 was a bit lower when E. coli SHuffle T7 was cultured under autoinduction conditions than in conventional induction with IPTG; since the full size L1(C30 was present in about 40 % of the soluble fractions corresponding to autoinduction conditions (Fig. 6, B, lanes 1 and 6 vs. 2-5 and 7-10). Thus, E. coli SHuffle T7, grown in autoinduction conditions, was able to produce slightly higher amounts of soluble truncated full size L1(C30 than E. coli BL1(DE3).
 A: Immunodetection of L1(C30 in whole cell lysates. Lane 1, pET28a Conventional IPTG (c) 125 µM; lane 2, pETHPV18L1(C30 c 125 µM; lane 3, pET28a auto-induction lactose (A-L); lane 4, pETHPV18L1(C30 A-L; lane 5, pET28a auto-induction IPTG (A-IPTG) 125 µM; lane 6, pETHPV18L1(C30 A-IPTG 125 µM; lane 7, pETHPV18L1(C30 A-IPTG 60 µM; lane 8, pETHPV18L1(C30 A-IPTG 30 µM. B: Soluble production of L1(C30 by E. coli SHuffle T7 [pETHPV18L1(C30]. Lane 1, c 125 µM soluble fraction (sf); lane 2, A-L sf; lanes 3, 4 and 5: sf A-IPTG 125 µM, 60 µM and 30 µM, respectively, lane 6, c 125 µM insoluble fraction (if); lane 7, A-L if; lanes 8 9 and 10, if A-IPTG 125µM, 60 µM and 30 µM, respectively.
A: Immunodetection of L1(C30 in whole cell lysates. Lane 1, pET28a Conventional IPTG (c) 125 µM; lane 2, pETHPV18L1(C30 c 125 µM; lane 3, pET28a auto-induction lactose (A-L); lane 4, pETHPV18L1(C30 A-L; lane 5, pET28a auto-induction IPTG (A-IPTG) 125 µM; lane 6, pETHPV18L1(C30 A-IPTG 125 µM; lane 7, pETHPV18L1(C30 A-IPTG 60 µM; lane 8, pETHPV18L1(C30 A-IPTG 30 µM. B: Soluble production of L1(C30 by E. coli SHuffle T7 [pETHPV18L1(C30]. Lane 1, c 125 µM soluble fraction (sf); lane 2, A-L sf; lanes 3, 4 and 5: sf A-IPTG 125 µM, 60 µM and 30 µM, respectively, lane 6, c 125 µM insoluble fraction (if); lane 7, A-L if; lanes 8 9 and 10, if A-IPTG 125µM, 60 µM and 30 µM, respectively.Fig. 6 Production of L1(C30 by E. coli SHuffle T7 in autoinduction conditions.
DISCUSSION
In Cuba, several HPV genotyping studies have been conducted to estimate the prevalence of oncogenic types in female Cuban patients with different grades of cervical lesions and cancer.23,24,27 In those studies, most individuals were infected with high-risk HPV type. Particularly, the HPV-18 type was among the three most prevalent genotypes. These findings support the future inclusion of a preventive HPV vaccine in the Cuban National Program of Immunization.23,27) Considering these facts, we decided to study the expression of a wild-type HPV-18 L1 gene in E. coli, which is an efficient and inexpensive platform with potentials to produce proteins with self-assembling properties.13,43
In this study, we have successfully isolated a HPV-18 L1 gene from a female Cuban patient and cloned it into E. coli. Analysis of its nucleotide sequence identified it as HPV-18 L1 gene and corroborated the previous diagnosis as HPV-18-positive. The cloned HPV-18 L1 gene was 99.9 % similar to the African variant EF202152, representative for HPV-18 sublineage B3, suggesting that the cloned L1 gene probably shares a common origin with the B lineage of genotype 18.
Taking into consideration that the expression of the full-length HPV-18 L1 gene has not been previously reported in E. coli, in the present study the full-length 18 L1 gene was subcloned into the plasmid vector, pET26b, and its expression was evaluated in E. coli BL21(DE3) and E. coli SHuffle T7. The native HPV-18 L1 protein was produced at low levels in both E. coli strains, which was in agreement with the prediction of low translation initiation rate of the L1 gene. Subsequently, the effect of N- and C-terminal truncations on the production and solubility of the native HPV-18 L1 protein was assessed in E. coli. According to our results, double truncated L1 protein variants were produced at similar levels and solubility in both E. coli strains, compared to the carboxy-terminal-only truncated variant; which indicated that the carboxyl terminal truncation was responsible for increasing the HPV-18 L1 protein amount and solubility. This result was in agreement with previous works about the positive effect of the C-truncation on production and solubility of HPV-16 L1 protein in E. coli,21,44 and disagreed with the reported previously about the little or no effect of deletions of up to 30 C-terminal residues on stability or solubility of the HPV-16 L1 protein in E. coli.5 Unexpectedly, we did not find a positive effect of the amino terminal truncation on the HPV-18 L1 production and solubility, which was congruent with the reported recently by Akuzum et al about the no production of the wild-type HPV16 and HPV18 L1 proteins with N-terminal truncations in E. coli.45 These findings could be related to an uncompleted removal of the potential inhibitory elements present at the 5´ end of the cloned HPV-18 L1 gene, according to previous reports about the presence of inhibitory RNA elements in the HPV‑16 L1 coding region.46 Similar inhibitory elements have been found in the L1 coding regions of HPV-5, HPV-6, HPV-18, HPV-31, HPV-45 and HPV-56,47 as well as in L1 from canine oral papillomavirus.48 Consequently, the fusion of 20 codons, with a 6xHis tag included at the 5’ end of the full-length HPV-18 L1 gene, led to the highest levels of HPV-18 L1 protein (Histag-HPV-18L1) in E. coli, which agreed with the highest TIR values predicted by the RBS Calculator. This last result could be explained by a possible displacement of the putative inhibitory elements due to the inserted fragment. Precisely, the transcriptional elements are one of the critical factors considered during optimization of synthetic genes for a chosen host. Thus, the use of a synthetic gene, with an optimized synthetic ribosome binding site sequence and with such inhibitory elements removed could promote higher amounts of the HPV-18 L1 protein in E. coli.
When previous studies on the effect of HPV L1 truncations on its production and solubility are reviewed, it is notorious that they were carried out with HPV L1 genes of different nucleotide sequences, and it is well known that the DNA sequence used to encode a polypeptide can have dramatic effects on its expression.49 Codon identity, mRNA secondary structure and nucleotide composition of the genes markedly influence its expression level in prokaryotic hosts.50,51 Consequently, the effect of N-terminal and C-terminal truncations of the HPV L1 protein on its production and solubility probably needs more experimental work and is actually a topic under active research.45,52
Respect to the evaluated E. coli strains, BL21(DE3) produced the higher levels of the three truncated variants of HPV-18 L1 and Histag-HPV-18L1, when grown under conventional induction with IPTG. We previously tested E. coli Rosetta as host for the expression of HPV-18 L1 gene, considering the high content of rare codons in this gene, but the production levels of HPV-18 L1 protein were lower than the produced by E. coli BL21(DE3).34 We had also evaluated E. coli BL21(DE3)-CodonPlus as host for the production of Histag-HPV-18L1 protein, but levels were similar to E. coli BL21(DE3) (data not shown). However, solubility of the three truncated variants of HPV-18 L1 was increased to more than 50 % in E. coli SHuffle T7, cultivated under conventional induction with IPTG. Particularly, E. coli SHuffle T7 produced about three times more amounts of the HPV-18 L1 variant truncated only at the carboxy terminus L1(C30, when it was cultivated under autoinduction conditions and levels were comparable to those induced under conventional induction with IPTG by E. coli BL21(DE3). To our knowledge, this is the first report about the soluble production of HPV-18 L1 protein in an E. coli SHuffle strain. Since E. coli SHuffle T7 produced the higher levels of L1(C30 under auto-induction with lactose is possible to increase the amounts and solubility of this protein through optimization of the fermentation conditions such as: pH, temperature, dissolved oxygen and growth medium.53,54 The soluble fraction of E. coli SHuffle T7 might have a suitable amount of properly folded HPV-18 L1, considering that its cytoplasm is maintained in an oxidative state and contains the Disulfide bond C (DsbC).55 Thus, current experiments are in progress to purify the L1∆C30 protein from the soluble fraction of E. coli SHuffle T7, considering the potential higher quality of L1 protein in this fraction compared to Ib. In the future, other strategies should be evaluated such as: the use of a synthetic gene, with optimized codon usage for E. coli, with an optimized synthetic ribosome binding site sequence and with inhibitory elements removed. All this with the aim to contribute to increase the amounts of HPV-18 L1 protein in E. coli.
Conclusions
The HPV-18 L1 gene cloned in this study had the highest level of sequence similarity (99.9 %) to the African variant EF202152, the representative for HPV-18 sublineage B3, suggesting that the cloned L1 gene probably shares a common origin with the B lineage of genotype 18. E. coli BL21(DE3) produced higher levels of the three truncated variants of HPV-18 L1 and Histag-HPV-18L1 than E. coli SHuffle T7, when conventional induction with IPTG was used. However, higher amounts of the three truncated proteins of HPV-18 L1 (more than 50 %) were detected in the soluble fraction of E. coli SHuffle T7. The variant truncated only by the carboxyl terminus, L1(C30 was produced to similar levels and solubility than HPV-18 L1s truncated at both termini, which indicated that the carboxy-terminal truncation of the HPV-18 L1 had a major contribution to the production and solubility of the wild-type HPV-18 L1 protein in the two E. coli strains tested. In autoinduction conditions, E. coli SHuffle T7 produced about three times more amounts of the variant truncated only by the carboxyl terminus than in IPTG´s conventional induction and levels were comparable to those obtained under conventional induction with IPTG by E. coli BL21(DE3). Thus, for the first time it is shown that E. coli SHuffle T7 strain produced the HPV-18 L1 protein in its soluble fraction. However, higher amounts of the L1 protein are needed to scale-up its production for developing a HPV vaccine candidate.

