










 Servicios personalizados
Servicios personalizados Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCTION
Lansberg's hognose viper (Porthidium lansbergii hutmanni) snakebite is an important medical emergency in Margarita and Coche Islands (Nueva Esparta state, Venezuela), a touristic destination located in the Caribbean Sea. The aftereffects are potentially serious, particularly if the patient is not treated fast and appropriately. Cornejo-Escobar et al. (2013)1 mention that in the country, Nueva Esparta state is not cited, as an important epidemiological area for ophidism. Nonetheless, De Sousa et al. (2013)2 indicate there were 328 ophidic accidents in this state from 1996 to 2004. Nowadays, the epidemiological importance of Porthidium l. hutmanni (P.l.h) and Crotalus durissus cumanensis is unknown (the two Viperidae species described in that geographical region) since the accidents in Margarita Island are classified as ophidism, without specifying the involved species.1
Porthidium l. hutmanni, locally known as “mapanare margariteña” is found in xerophytic semiarid forest and piedmonts in Margarita Island. Experimentally, P.l.h can produce oedema, necrosis, and bleeding (skin bruising, gastrointestinal haemorrhage, haematuria and lymphatic vessel damage with degradation of extracellular matrix effects).3,4 Fortunately, there have not been reported deaths from P.l.h bites in Venezuela.2 However, a local inflammatory response with bled through the lesion punctures, segmental oedema, and erythema associated to the touch was described in a published case.1
Previously, some biochemical and pharmacological characterization of the Porthidium venom has been experimentally done.3,4,5,6,7 Several fractions with haemorrhagic and proteolytic activity were described. The purification and characterization of a metalloproteinase (Porthidin-1), which had a potent activity in mice was carried out.4 Other studies, demonstrating activities on the platelets and causing haemorrhages and kidney damages have contributed to elucidate some physiopathological mechanisms induced by this venom.1,5,6,7
The main aim of this work was to describe the venom´s haemostatic characteristics as well as the potency appraisal of the polyvalent antiophidic serum (PAOS) against the haemotoxicity fromP.l.h experimental envenoming.
METHODS
Reagents
Electrophoresis: Reagents (BIO-RAD, USA), IPG Strips pH 3-10, 11 cm (BIO-RAD, USA); haemostasis: human fibrinogen (SIGMA, Mo, USA), bovine thrombin, benzamidine and ethylene-diamine-tetraacetic acid (EDTA) (SIGMA, Mo, USA); Immunoblotting: Equine peroxidase-coupled-equine IgG antibody (Santa Cruz Biotechnology, CA, USA), Nitrocellulose membrane (BIO-RAD, USA), SuperSignal West Pico® chemiluminescence development kit (ThermoScientific, USA); MALDI-TOF/TOF: α-Cyano-4-hydroxycinnamic acid matrix (α-CHCA) (SIGMA, Mo, USA), Acetonitrile, Trifluoroacetic Acid and diethyl ether (SIGMA, Mo, USA); LC-MS / MS: OFFGEL RoomTemp HighRes® Kit (Agilent Technologies, USA), IEF pH 3-10 24 cm strips (GE Healthcare, USA), Swine Trypsin (PROMEGA), Electro spray calibrate solution 63606 and Calibration Tune Mix ESI (SIGMA-Fluka, USA ); working solutions: reagents of high purity ≥ 98 % (Merck and Riedel de Haen, Germany). Polyvalent antiophidic serum (PAOS) was obtained from Biotecfar C.A, Caracas, Venezuela.
Software
For the statistical analyses the Prism® program (GraphPad, Software) was used. For the one dimension gels analysis the QuantityOne® (BIO-RAD, USA) software was utilised; while for the two-dimensional gels electrophoresis analysis the PDQuest® program (BIO-RAD) was employed.
Experimental animals
Male mice NIH strain weighing 20 to 22 g were handled for the acute toxicity determinations and mice from 25 to 27 g for evaluations of haemorrhagic activity. These animals were purchased from the National Institute of Hygiene "Rafael Rangel" (Caracas, Venezuela) animal facility. They were kept in cages under room temperature conditions, 12 h natural light with water and food at libitum until used.
Venom
Porthidium l. hutmanni venom pool was obtained by manual milking of 11 adult, both sexes specimens captured throughout the year in Margarita Island, Nueva Esparta state (Venezuela), located 2 to 30 meters above sea level. The Island coordinates are between 10.9971° North latitude and 63.9113° West longitude. The region of origin of the specimens under study presents a climate influenced by the northeast trade winds, with average annual temperatures of 28 °C and rainfall from 50 to 500 mm; the vegetation is xerophytic and semi-deciduous forests, its origin can be located in the tertiary geological era; forming part of the great Andean-American folding.8Bothrops colombiensis venom pool was from 12 adult, both sexes’ snakes, captured in different country locations. All specimens were maintained in captivity in the Serpentarium, Pharmacy School at the Universidad Central de Venezuela. The venom, once obtained, was crystallized under vacuum in a desiccator containing CaCl2 as a desiccant, and maintained at 4 °C, until used.
Fractionation of Porthidium l. hutmanni crude venom by molecular filtration
Fractionation of the P.l.h crude venom was started with a Sephadex® G-100 molecular exclusion chromatography column by Grillo and Scannone (1976)9 method. Venom fractions were dissolved in 5 mL of mobile phase, comprised of 0.2 M ammonium acetate (CH3COONH4) pH 6.8 (four runs were made, for a total of 1 000 mg of venom). Protein elution was accomplished by mobile phase isocratic gradient, at a flow rate of 7 mL/h. The eluates were monitored at 280 nm. Fractions were lyophilized, weighed and kept at -20 °C until estimating their haemorrhagic action.
A single dose of 1μg contained in 0.1 mL of the FI and FII fractions, in 0.85 % saline solution was inoculated in experimental animals, using four animals per group to define which fractions obtained by molecular exclusion had the highest haemorrhagic activity. The haemorrhagic area was established as described for the determination of the minimum haemorrhagic dose (MHD).
SDS-PAGE analysis of venom
Polyacrylamide gel electrophoresis (12 % SDS-PAGE) was carried out following Laemmli (1970) method.10 Briefly, samples were dissolved at a concentration of 5 μg/μL in a protease inhibitors cocktail, composed of 4-(2-aminoethyl)-benzene-sulfonyl fluoride (AEBSF), E-64, bestatin, leupeptin, aprotinin and ethylene-diamine-tetraacetic acid (EDTA); then diluted to the optimum concentration for visualization (crude venom: 2 μg/μL and FI: 1 μg/μL), in 0.5 M Tris-HCl buffer, pH 6.8, 10 % sodium dodecyl sulphate (SDS), 1 % glycerol and 0.02 % bromophenol blue. Five microlitres (5 μL) aliquot of the samples and 5 μL of pre-stained markers of wide molecular weight range were distributed into the gel. Once loaded in the respective wells of the gel, the samples were subjected to the electric constant current of 50 V, for 15 min, to allow the entry of the samples into the separation gel, and subsequently to constants 100 V until completing the run (about 120 min). Afterward, the relevant gels selected for staining were placed in a solution containing 12 % v/v phosphoric acid, 10 % w/v ammonium sulphate, 20 % v/v methanol and 0.12 % w/v Coomasie G-250 blue, according to the Blue Silver staining protocol,11 which has a sensitivity of 1 ng per band. Following, the gels were washed with deionized water to remove excess dye and digitised by densitometry. Each experiment was performed in duplicate.
Lethality: Lethal Dose Fifty (LD50)
The LD50 was determined by intraperitoneally (i.p.) venom injection in mice (18-22 g) and calculated according to the Spearman-Kärber method (1978).4 Five mice per dose (1.0 to 3.0 mg/kg) were used.
Determination of minimal haemorrhagic dose (MHD)
To determine MHD for the P.l.h crude venom and fractions, the modified Kondo (1960) method12 was used. Serial doses of venom or fraction (FI) in the range of 0.75 μg to 6 μg for crude venom; 0.075 μg to 2 μg for FI fraction were intradermal injected into the depilated backs of 20 mice (10 for crude venom and 10 for FI fraction). Two control mice injected with 0.85 % saline were used.The animals were then sacrificed and the skin removed after 2 h. The haemorrhagic diameter on the skin was measured and the MHD was defined as the amount of venom protein that caused a 10 mm haemorrhagic dot.
With the experimental data, the MHD was estimated by a dose response graph using the logarithm in 10 base values of the doses, for the abscissa axis, and the values corresponding to the diameters of the haemorrhagic lesions for the ordinates axis. Linear regression analysis was performed employing the Prism® program (GraphPad). This procedure was repeated in triplicate and the MHD mean and standard deviation were calculated.
Procoagulant Activity determination on Human Plasma
The ability of P.l.h crude venom and fractions to activate blood coagulation was determined by Theakston and Reid (1983) method.13 Concisely, different venom or fraction dilutions were prepared in a coagulation solution composed of 0.02 M phosphate-saline buffer solution (PBS) pH 7.4. Aliquots of 50 μL of each crude venom and FI dilutions were added to 200 μL citrated human plasma (from the laboratory member’s healthy donors). Regarding the crude venom, concentrations ranging from 0.1 μg to 100 μg per 50 μL; while for FI fraction, the concentrations range were from 10 to 300 μg. The coagulation time was recorded. Samples that induced plasma coagulation in less than 30 min were considered procoagulant.
Four duplicates were made for each dilution. Simultaneously, four negative control tubes were carried out, adding 50 uL of the coagulation solution instead of crude venom or fraction. In addition, incubation of 5 μg of Bothrops colombiensis venom and 200 μL of plasma (per quadrupled) was used as a positive control. In this case, the coagulation time should not exceed 60 s.
Anticoagulant determination activity
In addition to the previous experiment, attempts were made to determine whether the venom or the crude fractions of P.I.h, instead of coagulating the human plasma, inhibited their coagulation when recalcified. The modified method proposed by Rey-Suárez et al. (2011)14 was used. Succinctly, the procedure consisted of dilutions of venom in coagulation solution, in order to contain the required dose in 50 μL of solution; doses ranging from 0.1 μg to 100 μg were used. An aliquot of 50 μL of each dilution was added to 200 μL of citrated plasma, incubating at 37 °C for 10 min. During this period it was observed if plasma coagulation occurred. If not, 100 μL of 1M CaCl2 was added to the tube and again placed in incubation, observing for another 30 min, recording the coagulation time. Four replicates were made for each trial. The experimental control consisted of 50 μL of sterile 0.85 % saline, incubated with plasma in the absence of venom.
Defibrinating activity of Porthidium l. hutmanni crude venom and FI determinations
The venom or fractions in vivo proficiency to consume fibrinogen was assessed according to Gené et al. (1989) procedure.15 Treated five 20-22 g mice groups were intravenously inoculated with different dilutions of P.l.h crude venom or FI fraction, prepared in 0.2 mL sterile 0.85 % saline. With crude venom, doses ranging from 7.5 μg to 120 μg were inoculated; for FI fraction, the dose range was 6.25 μg to 100 μg. One hour after the inoculation, blood was drawn from the axillary plexus of each experimental anesthetised animal. Samples were kept in glass tubes for 2 h at room temperature. After the incubation time had elapsed, clot formation was observed. The minimal defibrinating dose (MDD) was defined as the minimum amount of venom that induced incoagulability in all inoculated mice. In addition, a control group was intravenously inoculated with 0.2 mL of 0.85 % sterile saline.
Determination of Porthidium l. hutmanni crude venom fibrinogenolytic activity
To evaluate the proteolytic activity of the P.l.h crude venom on the α, β, and γ chains of fibrinogen, the Gay et al. (2005) modified protocol16 was used. Briefly, a purified human fibrinogen stock solution was prepared at a concentration of 2mg/ mL in Tris-HCl 0.1M buffer solution, pH 7.4. Different concentrations of P.l.h crude venom (0.03 μg to 2 μg) and FI (0.03 μg to 4 μg), in a volume of 50 μL, were incubated at 37 °C for 30 min, with aliquots of 50 μL (100 μg) of the pre-prepared fibrinogen solution. After the incubation, each sample was diluted 1:1 with reducing solution containing: 0.5M Tris pH 6.8, 10 % SDS, 1 % glycerol, 0.02 % bromophenol blue, and 3 % of 2-β-mercaptoethanol. Then, samples were placed in water bath at 100 °C for 5 min. An aliquot of 15 μL of each sample was electrophoretic run as indicated. The action, on the different fibrinogen chains was evidenced by its degradation, with respect to what was observed in a fibrinogen sample run under the same conditions, in the absence of P.l.h crude venom or fraction. The assay was carried out by triplicate.
Determination of fibrinogenolytic activity as a function of time
Once the lowest amount of P.l.h crude venom (0.25 μg) capable of completely degrading the Aα chain of human fibrinogen under the above-described conditions was established, the concentration was incubated with 100 μg of fibrinogen at 37 °C, during different incubation times: 30 s, 1 min, 5 min, 15 min, 30 min, 60 min, 3 h and 24 h. Subsequently, these samples were electrophoresed (SDS-PAGE) as indicated for the determination of fibrinogenolytic activity and compared to the electrophoretic pattern of a control sample, which consisted of the corresponding dose of crude venom incubated with 100 μg of fibrinogen and immediately subjected to the reducing solution action (Time 0).
Protease inhibitors effect on fibrinogenolytic activity
Constant amounts of P.l.h crude venom (1 μg) were incubated with 100 μg of human fibrinogen in buffer solution 0.02 M Tris-HCl pH 7.5, at 37 °C for 30 min. To evaluate the presence of fibrinogenolytic serine proteases, the determination was carried out by adding 2 mM benzamidine to the incubation mixture, whereas for the evaluation of metalloprotease activity, the determination was made in samples with addition of EDTA. Samples without protease inhibitors were used as controls. After the incubation time had elapsed, the samples were run on SDS-PAGE as indicated for the determination of fibrinogenolytic activity.
Determination of fibrinolytic activity
The capacity of the degrading fibrin capacity of FI fraction of P.l.h. venom was evaluated following the method by Marsh and Arocha Piñango, (1972).17 Concisely, 1.5 mL of 0.1 % fibrinogen solution in Imidazole-buffered sterile 0.85 % saline, pH 7.4 were added to Petri dishes fibrin plates (3 cm). Then, 75 μL of bovine thrombin, at a 10 U/mL concentration, containing 0.025M CaCl2 was spilled, in order to form a uniform fibrin layer. Afterward, 10 μL (1 μg/μL) of FI fraction venom was prepared in sterile 0.85 % saline as carrier and placed in the centre of the fibrin layer, then incubated for 24 h at 37 °C. After the incubation time had elapsed, the diameter of the lysis area on the fibrin surface was determined. Fibrinolytic activity was expressed as the diameter of the lysis area per microgram of venom (mm2/μg). Bothrops colombiensis venom was used as a positive control and sterile 0.85 % saline as a negative control.
Determination of Porthidium l. hutmanni crude venom effects on platelet aggregation
These effects on platelet aggregation were assessed by turbidimetry according to Da Silva et al. (2009) method.18 Briefly, to obtain Platelet Rich Plasma (PRP), blood obtained from laboratory member’s healthy donors was centrifuged at 190 g, at 20 °C for 15 min. After platelet counting, in order to get poor platelet plasma (PPP), an aliquot was subjected to a second centrifugation at 1 700 g for 15 min. The plasma used during the trials consisted of PRP whose concentration was adjusted with PPP, at 300 000 platelets/mL.
During each determination in an aggregometer (Crono-Log® 560, USA), aliquot suspensions of 500 μL were placed under agitation at 37 °C in a siliconized cuvette; 10 μL of different dilutions of crude venom (0.6 μg to 16 μg), prepared in sterile 0.85 % saline were added to each sample. After 4 min, aggregation agonists adenosin diphosphate (ADP) (10 μM), ristocetin (1.25 mg/mL), collagen (8 μg/mL) and thrombin (1 U/mL) were added. The aggregation curve was recorded for 8 min in all the assays. As a reaction control, the agonists were placed on the platelet suspension, without venom, replacing it with 10 μL of sterile 0.85 % saline. A dose response curve was prepared with the results obtained to determine the Inhibitory Concentration 50 (IC50) for each agonist, which was defined as the amount of venom capable of reducing platelet aggregation by 50 % with respect to control.
Antivenom (PAOS) efficacy dose (ED50) neutralising the haemorrhagic activity
Five groups of four mice were tested with a mixture of antivenom containing the title declared for Bothrops genus by the PAOS manufacturer (1 mL of PAOS must neutralize the activity of 2 mg of Bothrops venom) diluted with sterile 0.85 % saline. A stock venom solution was freshly prepared at 0 °C prior to use. For each group of mice, a set concentration of venom was mixed with different antivenom concentrations and incubated at 37 °C for 30 min and centrifuged at 3 000 r.p.m. for 10 min (to eliminate the antigen-antibody complexes formed). Each mouse was intradermal injected with 0.2 mL of venom/antivenom mixture, in which 10 MHD in 0.1 mL was administered per mouse. After 2 h post injection, the haemorrhagic lesion as described for the MHD was evaluated. The corresponding percentage value for each dose of diameters reduction of the haemorrhagic lesions, by comparison with the control was calculated. The ED50 was defined as the amount of PAOS capable of reducing the area diameter by 50 % of haemorrhagic lesion.
Neutralization and antigenic recognition assays of Porthidium l. hutmanni crude venom and FI fraction by the PAOS (Immunoblotting assays)
The PAOS reactivity was evaluated against the linear epitopes present in the P.l.h crude venom and FI fraction by immunoblot. In this determination, the gel obtained from a one-dimensional electrophoresis was incubated during 10 min in transfer solution (50 mM Tris-HCl pH 8.0, containing 380 mM glycine, 0.1 % SDS and 20 % methanol). Then, the gel was placed in a transfer chamber to pass the proteins from the polyacrylamide matrix to a nitrocellulose membrane. This process was carried out at 180 mA for 2 h.
After the transfer, the nitrocellulose membrane was blocked for 2 h at room temperature, with a 0.2 M PBS, pH 7.0, with 5 % w/v skimmed milk and 0.1 % w/v Tween 20. Subsequently, three washes were performed for 5 min each with 0.2 M phosphate buffer saline (PBS), pH 7.0 solution, with 0.05 % w/v Tween 20). The sample was then incubated during 90 min with PAOS diluted 1: 3 000 in blocking solution at room temperature. After the incubation time had elapsed, three five-minute washes were made to each membrane. Immediately after, it was added a secondary antibody anti-equine IgG (coupled to horseradish peroxidase), diluted 1:7 000 in blocking solution, and incubated at room temperature for other 90 min. The electrophoretic PAOS bands were identified using a chemiluminescence development kit. The resulting image was then analysed.
Statistical analysis
The MHD calculations and their neutralization were performed by linear regression analysis. The data were expressed as the mean ± standard deviation. For the differences between experimental groups, the comparison was made by using one-way Analysis of Variance (ANOVA), specifically employing the Dunett test, a statistical test that estimates as significant those results with an error probability < 0.01.
RESULTS
Size exclusion chromatography
The P.l.h crude venom was initially fractionated by size exclusion. Two hundred and fifty mg of protein was separated using a Sephadex® G100 column. Proteins were detected at 280 nm. Four protein peaks were obtained (data not shown). A total of four fractionations were collected and pooled for further fractionation. Each fraction obtained was pooled, lyophilised and kept at -20 °C until use. Two predominant fractions were achieved (FI and FII) and used for further purification. These fractions presented the largest areas in the chromatogram.
Fraction I (FI) had a haemorrhagic area of 22.29 ± 2.79 mm that was produced by the inoculation of 1 μg fraction, whereas, the FII did not produce any haemorrhagic lesion with this dose.
Fractions electrophoresis (SDS-PAGE)
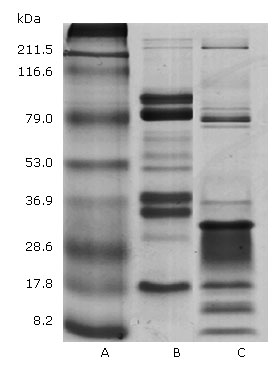
Figure 1 shows the P.l.h and Bothrops colombiensis venom electrophoretic profiles. In both examples, the distribution of protein bands occurred within gel regions corresponding to wide range molecular weights. In P.l.h crude venom 11 protein bands were evident. The high intensity bands accorded with the ∼79, 36.9 and 17.8 kDa molecular weights. Other lower intensity bands corresponded to ∼70, 60 , 53 and 32 kDa. While in the high molecular weight region only two low intensity bands higher than 211.5 kDas were evident. Regarding Bothrops colombiensis venom, 12 protein bands were obtained, in which the highest intensity corresponded to ∼79, 32, 17.8, 15 and 8.2 kDa, followed by weak bands of ∼ 215, 82 and 36.9 kDa were observed.
Lethality
Porthidium l. hutmanni crude venom inoculated intravenously in experimental animals showed a toxicity LD50 value of 2.51 mg/kg. The LD50 of the FI fraction was even lower than the P.l.h crude venom with a value of 1.45 ± 0.16 mg/kg, showing that this fraction possesses most of the venom toxic components.
Haemorrhagic activity
This test showed that the P.l.h crude venom MHD calculated was 1.47 μg. The purification factor was determined by the increase in haemorrhagic activity. A MHD of 1.47 µg was obtained from 1 000 mg P.l.h crude venom (yield= 100 %); the F1 fraction was 697.2 mg (yield= 69.7 %) with a MHD activity of 0.106 µg and a purification factor of 14.52.
Procoagulant activity
There was no plasma coagulation until 30 min by incubating samples of citrated human plasma with different amounts of venom. However, FI fraction of the P.l.h venom was able to induce human plasma coagulation, with a dose-dependent effect. The times recorded at all the doses tested were found to be much higher than those obtained against 5 μg of Bothrops colombiensis venom, in that case, for all the experimental replicates, the plasma coagulation time was 40 s.
Defibrinating activity
Porthidium .l. hutmanni crude venom intravenously inoculated did not produce blood incoagulability in mice blood, in any of the tested doses (data not shown). Hundred per cent of the experimental animals died, being the highest dose 120 µg/mouse; the experimental results showed that the mice blood treated with FI fraction, maintained its coagulant capacity after intravenous injection of every fraction, demonstrating lack of defibrinating activity.
Anticoagulant activity
Porthidium l. hutmanni crude venom was able to inhibit the citrated human plasma coagulation after its recalcification (it did not coagulate during the 30 min after the addition of 100 μL of 0.1M CaCl2. The plasma coagulation time (with 0.1 μg of venom), doubled with respect to the control value; the coagulation was totally inhibited when equal or higher doses than 1 μg were used.
Fibrinogenolytic activity
Porthidium l. hutmanni crude venom was able to degrade the fibrinogen α chain. This effect was dependent on the venom concentration after incubation for 30 min. The initiation of fibrinogenolytic activity from a venom/fibrinogen of 0.12 μg/100 μg and 0.5μg/100 μg ratios total degradation of the α chain was observed (Fig. 2).
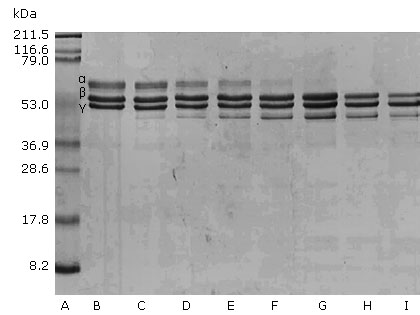
Porthidium l. hutmanni crude venom fibrinogenolytic activity in time function
Figure 3 represents the electrophoretic patterns of 100 μg of fibrinogen samples incubated with 0.25 μg of crude venom and exposed during different time intervals. The venom fibrinogenolytic activity was dependent on the incubation time. After 30 s of incubation at 37 °C, it was possible to demonstrate the fibrinogenolytic effect on α chain. After 30 min of incubation, the complete degradation of this chain was observed. By prolonging the incubation time to 24 h, it was determined that the venom also completely degraded the fibrinogen B chain. Degradation of the γ chain was not observed.

Effect of protease inhibitors on the Porthidium l. hutmanni crude venom fibrinogenolytic action
The analysis of the electrophoretic pattern of the fibrinogen chains treated with crude venom previously incubated with serine or metalloprotease inhibitors, revealed a clear inhibition of the venom proteolytic effect on fibrinogen by EDTA. No differences were observed with samples treated with benzamidine, with respect to the fibrinogen samples treated with crude venom without protease inhibitors (data not shown).
Fibrinolytic activity
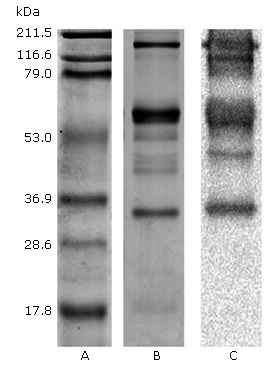
Porthidium l. hutmanni F1 fraction venom (6.95 ± 0.6 mm2/μg of venom) caused a degradation halo on the fibrin layer, which was significantly lower than the obtained by Bothrops colombiensis venom (24.98 ± 0.8 mm2/μg (p< 0.01) used as positive control (Fig. 4).

Activity of Porthidium l. hutmanni crude venom on platelet aggregation
Porthidium l. hutmanni crude venom inhibited platelet aggregation (IC50), in a dose-dependent manner induced by collagen (2.20 µg/mL), thrombin (6.06 µg/mL), ADP (3.97 µg/mL and ristocetin (1.29 µg/mL).
Neutralization of the Porthidium l. hutmanni crude venom haemorrhagic activity by PAOS
For the reason that PAOS does not declare a title of activity against the Porthidium genus venom, during the performance of these experiments, it was considered the B. colombiensis title, which neutralised ∼ 2 mg of venom per mL of PAOS.
The diameter reduction % of the haemorrhagic venom lesions (ED50) compared to the control group/mice, experiencing a decrease of approximately 9.0 % when incubated with 0.5 µg/µL PAOS; 38 % with 1 µg/µL; 33 % with 1.5 µg/µL; while, when exposed to 2 µg/µL, the P.l.h venom completely lost its haemorrhagic ability. The Bothrops colombiensis venom (56 μg) used as a control, against different dilutions of PAOS, resulted in 100 % neutralization of its haemorrhagic activity.
The PAOS antigenic recognition of Porthidium l. hutmanni crude venom by western blot
Figure 5 reveals the antigen-antibody recognition that occurred during the western blot performed between the P.l.h crude venom and the PAOS. Therefore, 5 of the 8 bands from P.l.h FI fraction venom proteins observed by SDS-PAGE were western blot PAOS recognised, with the exception of two bands corresponding to ~ 50 and 17.8 kDa molecular weights.
DISCUSSION
Porthidium l. hutmanni venom used during this determination, demonstrated that inoculated intravenously was very lethal, with a LD50 value similar to other reports using the intraperitoneally route.4,7 The venom and FI fraction showed local haemorrhagic production in the animal’s skin. The MHD value was higher than the before described.4 This is possibly due to the wide intraspecies variability proposed for snake venoms.19,20,21 The present study highlights this venom as the most haemorrhagic among the Venezuelan snake venoms that have been described.19,20) It is also among the most haemorrhagic species of Central and South America.
The presence of metalloproteases in P.l.h venom has been previously reported,4 these authors purified a metalloprotease of class P-1 called porthidin-1, showing slight haemorrhagic activity, this led to suggest that other metalloproteases could coexist in the venom.4 During the current study, several metalloproteases were demonstrated by proteomic techniques (data not shown), suggesting that it is probably the synergistic action of different snake venom metalloproteases (SVMP), particularly of class P-III, responsible for its high haemorrhagic activity.22 Recent proteomic analyses of snake venoms showed that metalloproteases were the major components in most of the Viperidae venoms; other toxic activities attributed to these enzymes have fibrinogenolytic actions, prothrombin and Factor X activation, induction of apoptosis, an inhibition of platelet aggregation, a proinflammatory activity, and an inactivation of serine protease blood inhibitors.23
Unlike the procoagulant effect, widely described for Bothrops venoms,19,24,25 and other included species within the "Porthidium Group" phylogenetic clade,26 for instance Atropoides nummifer27,28 and Cerrophidium godmani29 the P.l.h venom, used in the present study, proved to lack such activity. This absenc,e of coagulating capacity in Porthidium group venom has been previously reported in Atropoides picadoi, (15P. nasutum, (15,27,29P. ophryomegas, (15,29P. lansbergii lansbergii, (30 and P.l.h. (4
The lack of procoagulant activity in Porthidium spp. mainly points out to the absence of coagulation promoting substances. For example, Factor Xa analogs and enzymes with thrombin-like activity, described for Bothrops colombiensis venoms,31Bothrops isabelae32 and Bothrops atrox,33 or in the presence of proteins with anticoagulant action of such power, that allows to mask the plasma coagulant action, caused by the procoagulant present toxins.
The P.l.h venom anticoagulant action was able to prolong the human citrated plasma coagulation time (Howell time) after its recalcification, even at very low concentrations. This anticoagulant action by crude venom has not been reported for any Bothrops species, although some proteins with anticoagulant action have been isolated from this venom.34 However, to date, the components of the anticoagulant effect in different Porthidium species venoms remain to be elucidated.
Among the toxins of snake venoms, associated with anticoagulant effect, such as phospholipases A2 (with and without catalytic activity on the hydrolysis of phospholipids), L-amino acid oxidases, metalloproteases with fibrinogenase activity, serine proteases activating protein C, proteins with structure similar to lectins, which act as inhibitors of IX and/or X factors have been reported.35
Otherwise as inhibitors of thrombin and/or prothrombin and with the so-called toxins of three fingers, the latter described only in venoms of the Elapidae family snakes have been also stated. (36,37
The presence of different forms of L-aminoacid oxidases and metalloproteases (data not shown) has been demonstrated by mass spectrometry in the P.l.h venom. Some of these forms may be involved in these effects, without ruling out the occurrence of other anticoagulant proteins, identified in this venom. But this study also showed that this venom did not possess the characteristic defibring activity of bothrophic venoms, even at high doses, which reached the lethality in experimental animals.25
It has been proposed that the defibrinating action in bothrophic venoms is the consequence of metallo and serine protease activities, which act on the coagulation cascade, leading to fibrinogen consumption and fibrin microcoagula formation at the intravascular level38 or the enzymes action degrading fibrinogen, and generating products that are unable to polymerise.39,40,41
The haemostatic P.l.h venom effects found in this work clearly indicate that there were precise haemostatic differences between Porthidium and Bothrops venoms. Therefore, the classification in terms of the envenomation severity for P.h.l venom leads to a clinical approach that should not be performed under the same hemodynamic criteria used against bothropic ones.
The P.l.h crude venom proteolytic activity showed very interesting and comparable results to several Bothrops venoms, which have been widely reported and attributed mainly to the presence of serine and metalloproteases. (19,42 With respect to other haemostasis aspects, the fibrinogen is an important protein with a central role in blood coagulation, which has been revealed as a substrate of various Viperidae, Elapidae and Crotalidae venom enzymes.36 Enzymes with proteolytic action on fibrinogen and/or fibrin have been generically classified as fibrinogenases. Within them, α and β fibrinogenases stand out, which preferentially (but not exclusively) degrade α or β chain of fibrinogen or fibrin without releasing fibrinopeptides A or B, therefore, they do not induce coagule formation.43
In the present study was possible to demonstrate that the P.l.h venom possesses lytic activity on α and β chains of fibrinogen. This action quickly started on the α chain, while it was only possible to show an effect on the β chain by prolonging the incubation time, which indicated a fibrinogenases higher affinity for the human fibrinogen α chain, suggesting that this fibrinogenolytic activity could occur mainly due to this venom metalloproteases actions. This hypothesis is supported by analysing the protease inhibitors results on this activity; the fibrinogenolytic venom action was completely abolished by the metalloprotease inhibitor EDTA and not by benzamidine. This last potent serine proteases inhibitor did not show any inhibitory proteolytic action on venom upon the fibrinogen molecule. Similar results have been reported for Bothrops colombiensis,4 Crotalus durissus cumanensis20 and Micrurus tener tener44 venoms.
The P.l.h venom has demonstrated the degrading capacity on fibrin polymerized plates; however, when comparing this result with B. colombiensis assays in the current work, the latter has showed a powerful activity that was within the range of values previously reported for this species (16.8 ± 3.2 to 30.6 ± 5.5 mm2/mg);31 this fibrinolytic activity has been attributed mainly to the SVMP action,31 while, for P.l.h venom, the obtained value was approximately 3.6 times lower than that obtained for B. colombiensis.
The P.l.h venom effect on platelet aggregation was also evaluated. It is known that platelet thrombus formation is a fundamental event, in the maintenance of hemodynamic conditions, after the endothelial vascular rupture. Numerous snake venoms proteins, some with enzymatic activity, act as platelet activation and aggregation modulators, behaving in some cases as activators and in others as inhibitors of these processes.45
During the current study, it was demonstrated that the P.l.h venom had a powerful inhibitory effect against the ristocetin action, suggesting that it is capable of inhibiting the binding of von Willebrand factor (vWF) to the platelet receptor GPIbα. There are several mechanisms that have been associated with this effect, for example, vWF proteolysis by Class III metalloproteases such as jararagin (Bothrops jararaca), kaouthiagin (Naja kaouthia), and atrolysin A (Crotalus atrox);46 also, the GPIbα receptor proteolysis by toxins with activity similar to ~ 55kDa metalloprotease mocarhagin, isolated from the Naja mocambique venom, which breaks the peptide bond with high specificity between Glu282 and Asp283 residues of the GPIbα receptor; (47 the inhibition of GPIbα after its binding with C-type lectin-like proteins, such as equicetin (Equis carinatus) inhibiting the binding of vWF to the platelet surface.45
Here, the enzymatic activity of many bands decreased after incubating the P.l.h venom with EDTA, demonstrating the presence of metalloproteases in the tested venom. After reducing samples with 2-mercaptoethanol, no bands of high and low molecular masses could be seen, expressly in P.l.h venom. However, in the B. colombiensis venom, many bands were observed in these molecular weight locations. Important components of Bothrops genus venoms have been shown to be positioned in this gel region, such as proteins with myotoxic,48 coagulant activity, phospholipase A2 and metalloproteases with fibrinolytic and haemorrhagic49 actions.
We initially characterised the protein profiles of the two venoms by one-dimensional SDS-PAGE gel electrophoresis (Fig. 1). The venom composition of the two Viperidae species displayed a wide range of molecular weight proteins, from ~8 kDa to ~212 kDa in size. Remarkably, several protein bands appear to be conserved in the venom of the two species, although there also seems to be an important degree of inter-specific variation in toxin composition, withB. colombiensiscontaining the most complex profile andP.l.h.the most simple (Fig. 1). Antigenic cross-reactivity between FI fraction P.l.h venom was observed using equine polyvalent antivenom. Immunoblotting these separated venom proteins with equine polyvalent antivenom (PAOS) revealed broad immunological cross-reactivity, a great number of immunogenic components were detected directly above 28 kDa in the fraction (F1) used to isolate the haemostatic components of the P.l.h venom (Fig. 2).
PAOS is effective in neutralizing the haemorrhagic and fibrinogenolytic activities induced by P.l.h venom. These data harmonise with those shown,41 who verified that PAOS pre-incubated with B. colombiensis and colombienases-1 before fibrinogenolytic in vitro assay neutralized this activity and also haemorrhagic lesions caused by the crude venom. Enzymes presenting diverse activities existing in Porthidium genus venoms may take action on a varied range of tissues, producing several local and systemic effects, such as severe muscle and tissue necrosis, vascular endothelial damage and coagulation disorders.3,4,7
In conclusion, P.l.h venom have demonstrated, with the previous trials and others described experimentally by several authors, to be a venom with high lethal, haemorrhagic, proteolytic and procoagulant activities, whose description will have an enormous utility among clinicians, who deal with these accidents in its geographical distribution areas. The future characterization of this variety of toxins will serve to obtain better antivenoms for the treatment of these accidents.

