










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El término clínico vasculitis incluye grupos de enfermedades consideradas entre las más difíciles e interesantes de la medicina. Asimismo, atraen la atención de clínicos y patólogos por el enigma que encierran y por el reto intelectual que significan sus etiopatogenias. En general, se reconoce la participación de un mecanismo inmunológico, cuyo órgano diana es el endotelio vascular.1
Las manifestaciones clínicas son muy variables, y están en relación con la presencia de lesiones inflamatorias y necrotizantes de vasos sanguíneos en diferentes sistemas orgánicos, que causan isquemia distal y/o hemorragia de los órganos o territorios afectados. La extensión y gravedad oscila entre un cuadro cutáneo autolimitado y un compromiso multivisceral potencialmente mortal.1
Desde la segunda mitad del siglo XX, se ha tratado de perfeccionar la nomenclatura de las vasculitis. Actualmente se utiliza la terminología adoptada en la Conferencia de Consenso Internacional de Chapel Hill, en 2012.2 Pueden ser primarias (según el tipo de vaso que de forma predominante se encuentre afectado: grande, mediano y pequeño), estar asociadas a enfermedades del tejido conjuntivo, infecciosas o neoplásicas.1 Este último consenso, como aspecto novedoso, añade la identificación de otras dos categorías de vasculitis: de vaso variable y de órgano único.2
La granulomatosis con poliangeítis (GPA) es una enfermedad sistémica compleja tipo vasculitis, que afecta a vasos de mediano y pequeño calibre. Se le conoce por el epónimo de enfermedad de Wegener, en honor a su primer descriptor, el patólogo alemán Friedrich Wegener.3,4 Esta entidad anualmente tiene una baja incidencia: 5 a 10 pacientes por millón de población (pmp), al igual que su prevalencia (24-157 pmp). Puede presentarse a cualquier edad, aunque es más frecuente entre la tercera y quinta décadas de la vida; no existe preferencia de género y es extremadamente rara en personas de color negro de piel.5
El diagnóstico de la GPA habitualmente se realiza mediante una combinación de manifestaciones clínicas y pruebas de laboratorio, en tanto que el tratamiento depende de varios factores que influyen en el desenlace, así como en la progresión de la enfermedad.3 Por lo general, se describe una tríada clásica, dada por inflamación granulomatosa de las vías respiratorias superiores e inferiores y una glomerulonefritis necrotizante segmentaria, aunque puede existir compromiso multisistémico, evidenciado a través de síntomas oculares, dermatológicos, musculoesqueléticos y del sistema nervioso periférico.5
La inmunopatogénesis de esta enfermedad aún no está clara, aunque el conocimiento ha experimentado un progreso sustancial en los últimos años, y se cree que está involucrada la inmunidad tanto celular como humoral. Además, los antecedentes genéticos y los factores ambientales pueden desempeñar un papel importante.5
Desde mediados de la década de 1980, los anticuerpos anticitoplasma de neutrófilos (ANCA) han mostrado su utilidad como marcadores serológicos en las vasculitis sistémicas y se han vinculado con la patogenia de la enfermedad.6 Su presencia fue demostrada en pacientes con glomerulonefritis segmentaria, hemoptisis y hemorragia pulmonar.7 Constituyen autoanticuerpos circulantes de tipo IgM o IgG, dirigidos contra determinantes antigénicos localizados en gránulos primarios de leucocitos polimorfonucleares y lisosomas de monocitos activados, identificados mediante inmunofluorescencia o técnicas cuantitativas (enzimoinmunoensayo-ELISA).6
La realización del presente trabajo se basó en el reconocimiento de la GPA como una de las enfermedades raras.8 Las autoras se propusieron como objetivo analizar el interesante caso de una paciente con GPA, con una presentación sistémica, cuyo diagnóstico fue difícil y representó un gran desafío. Paciente que mostró una sobrevida, determinada por el tratamiento impuesto, acorde con los datos descritos en la literatura internacional.
PRESENTACIÓN DE CASO
Paciente femenina, de 59 años de edad, color blanco de piel, con antecedentes patológicos personales de ser fumadora inveterada e hipertensión arterial con 10 años de evolución, tratada con captopril y clortalidona, 25 mg diarios de cada uno. Siete días previos a la hospitalización, comenzó con decaimiento, fiebre de 38-38,5 ºC, escalofríos vespertinos, náuseas, orinas turbias y hemáticas, detectándose en analítica de urgencia creatininemia elevada al momento del ingreso.
Ante el presunto diagnóstico inicial de infección del tracto urinario, se le indica cefotaxima 2 g/día (ajustada según función renal). Después del tercer día de tratamiento, los síntomas constitucionales mejoraron e inmediatamente, de forma progresiva, comenzó con signos de poliartritis bilateral y simétrica, púrpuras palpables simétricas localizadas en miembros inferiores y glúteos, nódulos subcutáneos en antebrazo y mano izquierda, fenómeno de Raynaud, odinofagia, disnea y hemoptisis franca, generándose en la paciente un estado de gravedad que motivó su traslado a la Unidad de Cuidados Intermedios.
Como dato de interés, al interrogatorio se señala la presencia, tres meses antes de la hospitalización, de poliartritis bilateral y simétrica, recibiendo tratamiento con dosis baja de prednisona (10 mg/día) por prescripción reumatológica. Se constató, además, afectación ocular, interpretada como hemorragia subconjuntival por Oftalmología, período en el cual conservaba función renal normal (creatinina sérica: 59 μmol/l).
Exámenes complementarios
Hemograma: Hb: 81 g/l; Hto. 23 %; VCM: 82 fl; HCM: 30 pg; CHCM: 332 g/l; leucograma: 18,5 x 109/l, segm. 0,87, linf. 0,12, mon. 0,01; coagulograma: normal.
Hemoquímica: glicemia: 4,2 mmol/l; creatinina: 362 μmol/l; urea: 29 mmol/l; ácido úrico: 506 mmol/l; colesterol: 6,02 mmol/l; Tg: 1,79 mmol/l; PT: 53,1 g/l; albúmina: 35,5 g/l; perfil hepático: normal.
Reactantes de fase aguda: VSG: 105 mm/h; proteína C reactiva: 29 mg/l; F. reumatoideo: > 500 u/ml.
Estudios inmunológicos: C3: 1,22 g/l; C4: 0,23 g/l; anti-DNA dc: 7,7 u/ml (VR: < 30 u/ ml); ANA: negativo; c-ANCA: 47,9 u/ ml (VR: < 5 u/ml).
Orina: proteinuria de 24 horas: VT: 1540 ml, P: 1,01 g/24 h; conteo de addis (8 horas): VT: 610 ml, diuresis: 1,27 ml/mto, densidad: 1008; P: 1,1 mg/mto, L: 60960/mto, H: 152600/mto, C: 0/mto; urocultivo: no crecimiento bacteriano.
Hemocultivo: no crecimiento bacteriano.
Ultrasonido abdominal: RD: 102 x 46,5 mm, parénquima de 12,6 mm; RI: 106 x 47 mm, parénquima de 12 mm. Ambos con contornos regulares, buena relación corticomedular, no litiasis ni dilatación del sistema excretor.
Tomografía axial computarizada (TAC) simple de tórax: engrosamiento pleural focal, apical izquierdo. Microhemorragias multifocales en región basal de lóbulo superior izquierdo y lóbulo medio derecho. (Fig. 1 y 2)
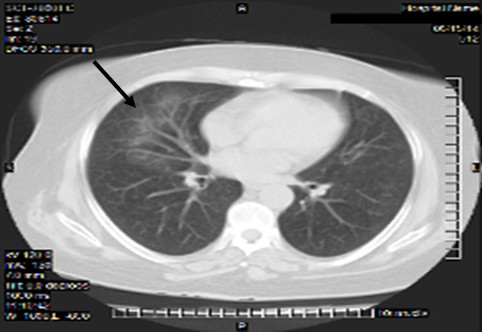
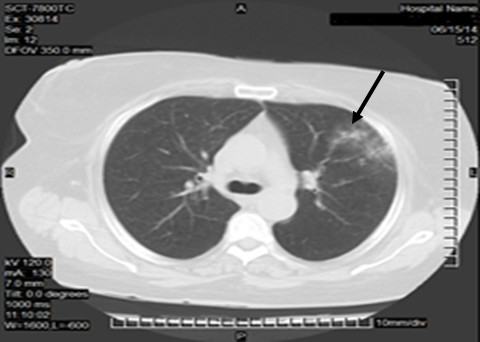
Ante la presencia del cuadro clínico anterior sugestivo de vasculitis sistémica, se comenzó tratamiento inmunosupresor potente. Fueron administrados tres pulsos de metilprednisolona (500 mg/día) seguidos de prednisona 1 mg/kg/día (70 mg/día) y bolos mensuales de ciclofosfamida 750 mg EV, lográndose control de la hemoptisis y mejoría paulatina del resto de los síntomas.
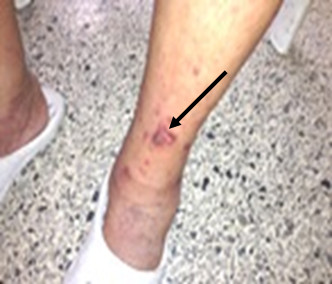
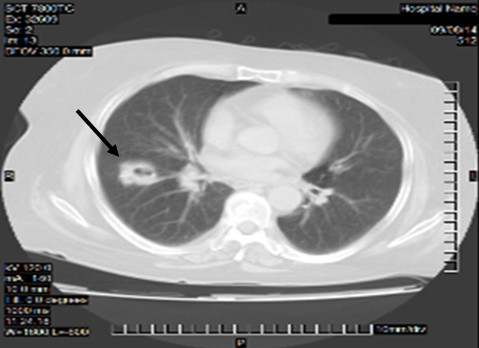
Evolutivamente, luego de tres meses de iniciado el tratamiento, desde el punto de vista respiratorio presentó tos con escasa movilización de secreciones, disfonía, lesiones cutáneas a nivel de nariz y extremidad inferior izquierda (vesico-ampollas y ulceración, figura 3) y desarrolló diabetes mellitus secundaria a esteroide. Desde el punto de vista renal, se constata mejoría en su función (descenso de creatinina sérica a 165 μmol/l) en los niveles de proteinuria (0,2 g/24h), y sedimento urinario solo con leucocituria, se normalizó VSG (22 mm/h). En radiografía de tórax y TAC simple de tórax evolutiva, figura 4, se constató imagen nodular de 30 x 28 mm de diámetro con radiotransparencia en su interior sugestiva de cavitación en lóbulo inferior de pulmón derecho, no existente en estudio anterior.
Después del octavo mes de cumplir adecuadamente terapia inmunosupresora, se constata empeoramiento clínico: los azoados elevan nuevamente sus niveles (creatinina en el orden de los 400 μmol/l) y se aprecian signos de expansión de volumen del líquido extracelular (anasarca, congestión cardiorrespiratoria), que motivaron el inicio de tratamiento sustitutivo de la función renal y la retirada paulatina de inmunoterapia. Ante la presencia de fallo renal irreversible, se mantiene en régimen de hemodiálisis iterada durante dos años y tres meses, falleciendo la paciente en su domicilio de manera súbita.
DISCUSIÓN
La vasculitis asociada a ANCA (AAV, por sus siglas en inglés) comprende un grupo de enfermedades autoinmunes multisistémicas, caracterizadas por serología ANCA positiva e inflamación de pequeños vasos. Las subcategorías de enfermedades comprenden GPA, poliangeítis microscópica, granulomatosis eosinofílica con oliangeítis y vasculitis ANCA limitada a riñón. La afectación renal ocurre en más del 50 % de GPA y en el 80 % de pacientes afectos de poliangeítis microscópica, frecuentemente presentándose como glomerulonefritis rápidamente progresiva. En los últimos años, los avances en el conocimiento de la fisiopatología de AAVs y el desarrollo de estrategias terapéuticas más efectivas, han conducido a mejoría significativa en los resultados; sin embargo el pronóstico a largo plazo continúa siendo subóptimo.9,10
La GPA y la poliangeítis microscópica han sido asociadas con un índice de supervivencia a los 5 años del 74-79 % y 46-80 %, respectivamente.11 La enfermedad renal crónica grado 5 ocurre entre el 20 y el 40 % de los casos.9
En el año 2022, un grupo internacional de expertos aprobado por el American College of Rheumatology y la European Alliance of Associations of Rheumatology7 desarrolló y validó los criterios de clasificación de las vasculitis asociadas con ANCA. Específicamente la GPA se define ante la presencia de:
Secreción nasal sanguinolenta, costra o congestión nasal (+3)
Afección de cartílago (+2)
Nódulos pulmonares, masas o cavitaciones en tórax por imagen (+2)
Conteo de eosinófilos ≥ 1 x 109/l (-4)
Anti-PR3-ANCA positivo: anticuerpos contra enzima lisosómica en neutrófilos (proteinasa 3), determinante del típico patrón citoplasmático (c- ANCA) (+5)
Glomerulonefritis pauciinmune (+1)
Antimieloperoxidasa-ANCA positivo (-1)
Granulomas o células gigantes por biopsia (+2)
Pérdida de audición conductiva o sensineural (+1)
Inflamación o consolidación de senos nasal/parasinusal por imagen (+1)
Se establece una puntuación para cada criterio, cuando esta es superior a 5, el diagnóstico es altamente probable, con una sensibilidad del 85 % y especificidad del 99 %.7
La sintomatología expresada por la paciente muestra la heterogeneidad en la forma de presentación de la GPA, aspecto que coincide con reportes de la literatura revisada por las autoras. Hernández Acosta et al.3 plantean que habitualmente el compromiso reportado en estudios clínicos consiste en afectación renal, síntomas constitucionales (fiebre, pérdida de peso, mialgias, entre otros) y manifestaciones otorrinolaringológicas. Tal como ha sucedido en otros pacientes, la respuesta no adecuada al uso de antimicrobianos y la ausencia de gérmenes en cultivo de orina y sangre, orientaron a que no se trataba de una entidad infecciosa.
El diagnóstico de GPA en esta paciente, que desarrolló de manera abrupta síntomas que pusieron en peligro inminente su vida por afectación de los aparatos respiratorio y renal, representó un gran desafío, ante todo por la urgencia de definir un tratamiento inmediato que impidiera un desenlace fatal. La evolución del caso condujo al planteamiento nosológico de una vasculitis sistémica y, como tal, fue tratada de forma inmediata. En el curso de la terapia inmunosupresora se tomaron las muestras correspondientes para estudios inmunológicos, obteniéndose como resultado c-ANCA: 47,9 u/ml (VR: < 5 u/ml), elemento que fue clave para el diagnóstico de GPA. En la poliangeítis microscópica y en la granulomatosis eosinofílica con poliangeítis los ANCA son mayormente dirigidos contra la mieloperoxidasa y determinan el patrón perinuclear de la enfermedad (p-ANCA).12
Tradicionalmente la biopsia ha constituido un método diagnóstico de elección. Histopatológicamente el pulmón es el órgano con mayor rendimiento diagnóstico; en el riñón suele encontrarse una glomerulonefritis segmentaria necrosante inmunonegativa con semilunas epiteliales, raramente se aprecian signos de arteritis o granulomas.13 En los últimos años y con el advenimiento de la dosificación de los ANCA, se ha simplificado el diagnóstico de esta enfermedad, ante el reto impuesto para preservar la vida de los pacientes.8
Los exámenes de imagen también son de vital importancia en el estudio de GPA y constituyen un pilar importante en los criterios diagnósticos. La tomografía computarizada es la técnica de elección para la caracterización de las manifestaciones en GPA, por su alta resolución espacial, amplia disponibilidad y menor costo. Presenta alto rendimiento diagnóstico en patología de cavidades paranasales, y es el examen con mejor relación costo/efectividad en la evaluación del tórax.14
En ausencia de tratamiento adecuado el pronóstico es fatal, con una supervivencia media de 5 meses tras el desarrollo de la afección renal y con una mortalidad del 90 % a los dos años. La introducción de efectiva terapéutica citotóxica inmunosupresiva ha cambiado de forma dramática el curso de la GPA, mejorando el pronóstico de los pacientes afectados.15
Las autoras de este trabajo consideran que en la paciente que se presenta, el debut dado por una forma generalizada de la enfermedad, con compromiso significativo de órganos vitales como pulmón y riñón, aparición de complicaciones propias de la GPA y secundarias al tratamiento médico, ensombrecieron el pronóstico en relación con la supervivencia prolongada. No obstante, la terapéutica impuesta hizo posible una sobrevida de dos años.

