










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La biomasa lignocelulósica es uno de los recursos orgánicos más abundantes a nivel mundial, y constituye una fuente prometedora para la obtención de energía renovable y bioproductos 1; es considerada una alternativa a las fuentes fósiles de carbono como el petróleo, gas natural y carbón, y tiene una importante aplicación en la producción de polímeros, fertilizantes, etc.2 Su composición varía según la fuente (por ejemplo: maderas duras, maderas suaves, residuos de la agricultura y cultivos energéticos), y está afectada por su origen, edad, condiciones climáticas, y por los procesos de cosecha y almacenamiento. Está conformada por tres componentes poliméricos mayoritarios que se encuentran entrelazados: celulosa, hemicelulosa y lignina, además de extractables y numerosos materiales inorgánicos.3
La celulosa y la hemicelulosa son polisacáridos que pueden convertirse en compuestos químicos a base de furanos y en ácidos orgánicos por vías termoquímicas. También pueden transformarse a bioetanol, biobutanol, biogás y otros productos de fermentación a través de varias vías biológicas. En los procesos de conversión biológica para la obtención de estos productos, diferentes propiedades de la biomasa como la composición química, el peso molecular de la lignina, la cristalinidad, el grado de polimerización y la accesibilidad de la celulosa, ocasionan su recalcitrancia.4
Se ha reportado que una delignificación de la biomasa reduce las barreras físicas y químicas, e incrementa la accesibilidad de la celulosa, con lo que se logra mejorar su bioconversión. También la modificación en la estructura de la lignina mejora la sacarificación enzimática de la biomasa, aun cuando no se logra una remoción significativa de la misma.4
Dichas modificaciones se pueden obtener al realizar un paso de pretratamiento previo a la etapa de hidrólisis, que puede ser químico, físico, físico-químico y biológico.5 Este proceso es principalmente utilizado para incrementar la producción de biogás y etanol; sin embargo, también puede emplearse para mejorar el rendimiento en la producción de todos los bioquímicos obtenidos a partir de lignocelulosas, así como en la obtención de oligosacáridos para la alimentación animal.6
La gran variedad de materiales lignocelulósicos hace difícil encontrar un diseño general del proceso para todas las materias primas. La recalcitrancia de las maderas suaves es mucho mayor que la de la mayoría de los cultivos y residuos agrícolas y herbáceos. Por lo tanto, la selección del “mejor método de pretratamiento” no es tarea fácil. Su selección depende en gran parte de la aplicación final, y cualquier recomendación debe basarse en una minuciosa evaluación técnico-económica en la que los datos hayan sido recolectados, al menos, a escala piloto.2
Uno de los desafíos en la selección del pretratamiento es evaluar el efecto de los diferentes métodos. Por ejemplo, si el propósito es producir etanol o biogás, se pueden emplear varios indicadores para evaluar el material pretratado, entre ellos la estimación de los niveles de compuestos tóxicos o inhibitorios que pueden causar un detrimento en procesos de hidrólisis enzimática y fermentación. Por otro lado, si el propósito es producir polímeros, geles o aglutinantes, otros criterios de evaluación deben ser empleados, como la fuerza mecánica y las propiedades de hinchamiento en el caso de los polímeros.2
El efecto del pretratamiento no solo depende de la biomasa empleada y el tipo de pretratamiento, sino de las condiciones que se utilizan (tiempo, temperatura, etc.), que constituyen también parámetros a optimizar. Este trabajo tiene como objetivo presentar una revisión de los principales métodos analíticos utilizados para la evaluación de las técnicas de pretratamiento empleadas en biomasas lignocelulósicas, así como otros métodos alternativos que pudieran ser empleados con este fin.
Desarrollo
Varios análisis han sido desarrollados para evaluar el efecto de pretratamientos aplicados a biomasas lignocelulósicas, y para describir las mejoras en los procesos de hidrólisis enzimática o microbiana 6, los cuales son necesarios en la obtención de productos como el biogás y el etanol. Entre los principales aspectos a tener en cuenta para ambos casos se encuentran los cambios en la composición, estructura, cristalinidad y formación de sustancias tóxicas e inhibitorias. En el caso específico de la obtención de biogás existen investigaciones que evalúan el grado de solubilización de la biomasa, expresado como la demanda química de oxígeno y los ácidos grasos volátiles (figura 1).7-8

Fig. 1 Principales análisis realizados a biomasas lignocelulósicas pretratadas para obtención de biogás. Fuente: Adaptado de Karimi y colaboradores 6
Específicamente para el estudio de la composición de la biomasa lignocelulósica se utiliza, fundamentalmente el método de Van Soest, basado en el uso de detergentes para fraccionar la fibra y conservar la lignina en el análisis de forrajes, los métodos para determinar parámetros en los procesos relacionados al sector del biogás propuestos por el Centro de Investigación de Biomasa de Alemania (German Biomass Research Center) y los Procedimientos Analíticos Estándares para Biomasa empleados en Estados Unidos 9, desarrollados estos últimos por el Laboratorio Nacional de Energía Renovable (National Renewable Energy Laboratory, NREL), quien ha desarrollado un conjunto de procedimientos analíticos para caracterizar la biomasa lignocelulósica e investigar los cambios composicionales, químicos y estructurales después de un pretratamiento, y su efecto en la hidrólisis enzimática, enfocados en la conversión de biomasa a biocombustibles (biogás/bioetanol). Estos procedimientos incluyen la determinación de ST, SV y cenizas, el contenido de proteína, azúcares y sus productos de degradación, y el análisis de carbohidratos y lignina.10
Sólidos totales, sólidos volátiles y cenizas
Las muestras de biomasa pueden contener cantidades grandes y variables de humedad, que pueden cambiar muy rápido cuando se exponen al aire, por lo que los resultados de los análisis de su composición química se reportan típicamente en base al peso de la muestra seca, que permite la comparación de muestras en una base consistente 10, además, se ha demostrado que el contenido de ST guarda relación con el rendimiento de biogás obtenido para diferentes sustratos.11
Los ST constituyen el residuo que se obtiene en el recipiente al evaporar a sequedad una cantidad de muestra determinada.12 Durante este procedimiento se elimina el agua de la materia fresca, aunque para sustratos con una fracción significativa de SV (como forrajes) se debe considerar la pérdida de compuestos orgánicos durante el secado.13 Se han realizado propuestas para corregir el contenido de ST en diversos materiales, como la remolacha azucarera, a través de ecuaciones que consideran el pH, el contenido de ácidos orgánicos, alcoholes de bajo peso molecular y ácido láctico.14 A temperaturas de 103-105°C, la pérdida de materia orgánica por volatilización es generalmente escasa, aunque sí puede influir en la sobreestimación del rendimiento de metano en procesos de digestión anaerobia. Angelidaki y colaboradores 15 recomiendan para materiales con alto contenido en AGV incrementar el pH para decrecer su volatilidad durante la determinación de ST o trabajar a 90 °C.
Al someter el residuo del análisis de ST a ignición a 550 °C, los componentes orgánicos se oxidan y se obtiene una fracción inerte conocida como cenizas. En muestras lignocelulósicas hay dos tipos de ceniza: no extraíbles y extraíbles. Las primeras son los materiales inorgánicos enlazados a la estructura; y las extraíbles, los que se adhieren a la biomasa y se eliminan por lavado con agua; su forma típica es el suelo.16 En lignocelulosas con alto contenido de cenizas (más de 10 %), el análisis de lignina y carbohidratos fibrosos propuesto por Sluiter y colaboradores 17 no es adecuado, pues pueden neutralizar una parte del ácido sulfúrico y afectar considerablemente los resultados analíticos.
Al sustraer la masa de la fracción inerte de la masa inicial de ST se obtiene el contenido de SV 18, que representa una aproximación de la fracción orgánica presente en el sustrato.13
Separación de las fracciones líquida y sólida
Al someter una muestra a pretratamiento, las fracciones sólida y líquida deben ser separadas para los análisis posteriores; esto puede lograrse por filtración a vacío o por centrifugación, donde se recomienda trabajar a velocidades de 6 000 rpm por 15-30 min.19 La FS ya separada debe ser lavada con agua o pequeñas cantidades de detergente, y volverse a filtrar, y luego se realiza el secado.20,21
Determinación de la composición química. Carbohidratos fibrosos
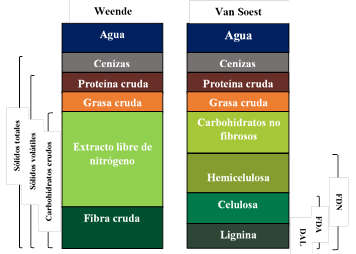
Varios investigadores han desarrollado aproximaciones para estimar la composición de diferentes sustratos. La determinación analítica se basa, típicamente en el análisis proximal (método Weender) y el método de los detergentes de Van Soest (figura 2).13
El sistema de análisis de Weender es un método gravimétrico que consiste en colocar una muestra secuencialmente en reflujo en base diluida (NaOH), seguido por ácido diluido (H2SO4), aunque se han realizado modificaciones en los últimos años que reportan el uso inicial del ácido y luego de la base, también diluidos al 1,25 %.22 El residuo resultante se conoce originalmente como fibra cruda, que actualmente se sabe que está compuesto principalmente por celulosa y proporciones variables de polisacáridos no celulósicos y de lignina. Según Weender, la FC representaba la fracción menos digerible de los alimentos, y el extracto libre de nitrógeno la de los carbohidratos más digeribles, lo cual conducía a resultados erróneos 23, aunque este método sigue siendo empleado en el análisis composicional de biomasas lignocelulósicas que son sometidas a pretratamiento 7-24, además de su uso en la evaluación de la calidad nutritiva y digestabilidad de los forrajes.25-26
 Fuente: Weinrich y colaboradores 13
Fuente: Weinrich y colaboradores 13
Fig. 2 Componentes del análisis de Weender y Van Soest de nutrientes característicos
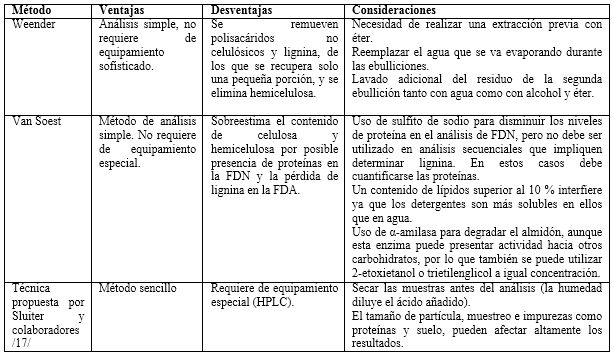
La propuesta de Van Soest se basó sobre el uso de detergentes, debido a la capacidad que tienen algunos (como el laurilsulfato de sodio) de solubilizar proteínas, así como la propiedad de algunos compuestos de amonio cuaternario (como el bromuro de cetiltrimetilamonio) para disolver polisacáridos, proteínas y ácidos nucleicos. Según Van Soest, estos compuestos servirían para fraccionar la fibra conservando la lignina. Van Soest declaró que de la FDN hacen parte la hemicelulosa, celulosa, lignina y otros componentes (combinaciones de nitrógeno con hemicelulosas y lignina con celulosa, proteína y otros). Para la FDA sus principales constituyentes serían los mismos que para la FDN, excepto la hemicelulosa (figura 2). Este método sigue siendo utilizado para el análisis de compuestos lignocelulósicos pretratados 24,27,28, aunque se deben tener en cuenta varios aspectos para llevar a cabo el análisis (tabla 1).29 También se ha reportado el uso de este método como una segunda etapa después de realizado el análisis de proteína y grasa cruda, carbohidratos no fibrosos y fibra cruda, determinados por el método de Weender en biomasas lignocelulósicas antes de someterse a digestión anaerobia.30,31
Otra de las vías para la cuantificación de carbohidratos estructurales y lignina es la propuesta por Sluiter y colaboradores 17, basada en una hidrólisis ácida de dos etapas. La biomasa se somete, en primer lugar, a hidrólisis con ácido sulfúrico (H2SO4) al 72 %, seguido de dilución de la mezcla para obtener hidrólisis con H2SO4 al 4 % a 120 °C en un recipiente sellado. Después la mezcla se neutraliza por encalado, y los azúcares simples y los grupos acetilo liberados son determinados por HPLC. En este método, la lignina se clasifica como fracciones AIL y ASL. El material soluble en ácido es la fracción de lignina que tiene un peso molecular bajo, la cual se solubiliza en la hidrólisis ácida y se determina por espectrofotometría UV-Visible. La lignina insoluble en ácido, por otra parte, es la de peso molecular elevado que no puede ser disuelta durante la hidrólisis y se mide gravimétricamente. Este tipo de lignina se determina por la filtración de los materiales después de la hidrólisis ácida, secado a 105 °C, y calcinado a 575 °C.17 Este método es sencillo y ha sido empleado por varios autores para evaluar la influencia de los pretratamientos en biomasas lignocelulósicas 32,24; sin embargo, se debe prestar especial atención para obtener resultados confiables (tabla 1).
Determinación de proteínas
Las proteínas constituyen una interferencia en la determinación de lignina; por lo tanto, para su determinación exacta en los materiales lignocelulósicos con cantidades considerables de proteínas, su cuantificación es esencial.35 En el método presentado por Sluiter y colaboradores 17, las proteínas no hidrolizadas dan lugar a una sobreestimación de la lignina. Estas pueden determinarse simplemente mediante la medición del contenido de nitrógeno (por ejemplo, por el método Kjeldahl) y luego se estima el contenido de proteína utilizando un Factor Nitrógeno apropiado (factor N).6 Se han publicado varios métodos que recomiendan el uso de un factor de 6,25 para todo tipo de biomasa, excepto en granos de trigo donde se recomienda un factor de 5,7 35; también se ha determinado el factor nitrógeno para otros tipos de biomasa. El nitrógeno determinado por este método es la suma de nitrógeno orgánico e inorgánico 36 y ha sido utilizado en la determinación de nitrógeno y proteínas para la evaluación de pretratamientos 19, aunque existen otras técnicas para determinar el contenido de proteínas, más comúnmente empleadas en bioquímica, como el método de Lowry, Bradford, BCA y otros.37
Determinación de carbohidratos no estructurales
Dos tipos de carbohidratos pueden estar disponibles en la biomasa: estructurales y no estructurales. Los carbohidratos que están unidos en la biomasa se llaman estructurales, y los que se pueden eliminar por extracción o lavado se llaman no estructurales. El método de Sluiter y otros 17 es adecuado para carbohidratos estructurales, pero no para los no estructurales. Por ejemplo, cuando el sorgo o caña de azúcar, con un poco de azúcar residual, se somete al análisis, los azúcares libres deben primero ser analizados y removidos.
Estos carbohidratos no estructurales son polisacáridos constituidos principalmente por subunidades monoméricas de glucosa, xilosa, arabinosa, galactosa y manosa. Durante ciertos pretratamientos de biomasa, una parte de estos polisacáridos se hidroliza y se liberan azúcares solubles a la FL.38
En la tabla 2 se muestran varios métodos utilizados para el análisis de monosacáridos y carbohidratos no estructurales en biomasas lignocelulósicas: refractometría, colorimetría, metanólisis, CG, HPLC, cromatografía gas-líquido, cromatografía de intercambio aniónico (AEC), cromatografía de intercambio aniónico de alto pH con detección de pulso amperométrico (HPAE/PAD), espectroscopía de resonancia magnética nuclear protónica (RMN-1H), de infrarrojo cercano (NIR), análisis termogravimétrico (TGA), y electroforesis capilar (CE).6 En la evaluación de las técnicas de pretratamiento se han utilizado fundamentalmente CG 19 y HPLC.39
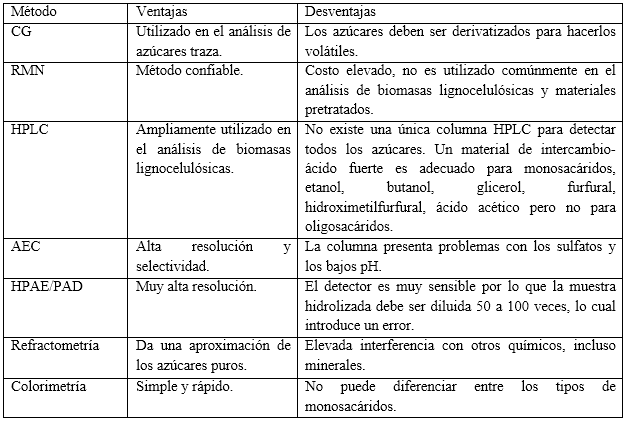
Sluiter y colaboradores 38 proponen un método para cuantificar la cantidad total de carbohidratos solubles, así como la cantidad de azúcares monoméricos liberados en solución. Los azúcares solubles en la FL de las muestras procesadas se pueden cuantificar mediante HPLC con detección del índice de refracción. Si los azúcares están presentes en forma oligomérica, se requiere un procesamiento adicional en sus unidades monoméricas antes del análisis. Los métodos de CG y HPLC son muy útiles para la determinación de monosacáridos en la FL, pero también son muy costosos y trabajosos, por lo que el uso de técnicas más sencillas como la colorimetría, constituye una alternativa para la cuantificación de carbohidratos no estructurales.
El método de fenol-ácido sulfúrico o de Dubois ha sido uno de los métodos más utilizados para la determinación de carbohidratos totales por espectrofotometría.40 Este se basa en la deshidratación de azúcares por acción de H2SO4 para formar furfural e HMF, que posteriormente reaccionan con el fenol para formar un compuesto coloreado que permite la cuantificación por espectrofotometría UV-Visible a una longitud de onda de 490 nm. Constituye un método sensible y ofrece resultados reproducibles, aunque investigaciones recientes han demostrado que no es necesario el uso del fenol, sino solo la hidrólisis con H2SO4 concentrado y la medición a 315 nm.41
El método de Miller es uno de los más empleados en la determinación de azúcares reductores debido a su bajo costo y toxicidad de los reactivos, alta sensibilidad y productividad, por lo que ha sufrido varias modificaciones a través de los años para adecuarse al análisis de diferentes materiales.40 Se basa en la reacción de los grupos reductores de los azúcares con el DNS. El DNS se reduce formando el ácido 3-amino-5-nitrosalicílico, mientras que el grupo aldehído se oxida para formar un grupo carboxilo. La lectura para la cuantificación se realiza a una longitud de onda de 540 nm.42 Este método ha sido empleado en la evaluación de pretratamiento alcalino en rastrojo de maíz.32
La cuantificación de los azúcares no reductores se puede determinar por diferencia entre los azúcares totales y los azúcares reductores.40 Estos métodos colorimétricos constituyen una alternativa al uso de técnicas más costosas y resultan rápidos y confiables.
Separación de los extractivos en fracción sólida
Los extractivos son una mezcla de diferentes productos químicos: resinas, proteínas, fitosteroles, grasas, ceras, sales, azúcares libres y una serie de hidrocarburos no volátiles en porciones menores. Estos compuestos interfieren en el análisis de lignina, por lo que deben extraerse primero para obtener resultados aceptables. Por ejemplo, la corteza de un pinar, la cual contiene un 19 % de extractivos, se analizó utilizando el método de Sluiter y colaboradores 43, con y sin la separación de extractivos.44 El contenido de lignina cuando los extractivos no se separaron fue superior en un 50 % respecto a cuando se realizó la extracción previa (material 6, tabla 3).
Tabla 3 Influencia de un paso de extracción en el contenido de lignina de la biomasa original (expresado como por ciento en base seca)

Fuente: adaptado de Burkhardt y colaboradores 44
Un método simple y estandarizado, sugerido en el Método Estándar de Análisis para la Determinación de Extractivos Etanólicos en Biomasa (ASTM E1690) y modificado por Sluiter y colaboradores 43, se utiliza ampliamente para el análisis de materiales pretratados para biocombustible. De acuerdo con este método, los extractivos son simplemente extraídos en un extractor Soxhlet por agua o un disolvente, generalmente etanol. Los materiales inorgánicos y nitrogenados, como ácidos de azúcares y azúcares no estructurales pueden ser extraídos por el agua, mientras que materiales cerosos y aceitosos pueden ser extraídos por etanol.43
Otra de las sustancias que interfiere en el análisis de compuestos lignocelulósicos es el almidón, que se hidroliza a glucosa en condiciones menos severas que la celulosa. Bajo las condiciones de hidrólisis presentadas por Sluiter y otros 17, el almidón se convierte en glucosa, pero una parte de esta se descompone en otros productos, como el 5-hidroximetilfurfural, por lo que se requiere una hidrólisis separada para el análisis de almidón, que puede ser por vía enzimática con α-amilasa 45; luego, el contenido de almidón puede ser restado del total de glucanos.
Determinación de la Demanda Química de Oxígeno Soluble
La Demanda Química de Oxígeno (DQO) se define como la cantidad de un oxidante específico que reacciona con la muestra en condiciones controladas. Los componentes orgánicos e inorgánicos de una muestra están sujetos a oxidación, pero en la mayoría de los casos predomina el componente orgánico.46 Similar a los SV, es una medida para los componentes del sustrato orgánico, pero generalmente se utiliza para la evaluación de muestras muy diluidas en el análisis de aguas residuales. Durante la digestión anaerobia de sustratos altamente diluidos con una alta proporción de sustancias volátiles (por ejemplo, filtración), también se utiliza la DQO, ya que ningún resultado se reporta en base a los ST.47
Cuando se realiza un pretratamiento, se determina la DQOs, que se corresponde con el valor de DQO de la FL, para evaluar su efecto en la solubilización de la materia orgánica. Su determinación se puede realizar por vía volumétrica o colorimétrica.
Ambos métodos se basan en la reacción de una cantidad de muestra con un oxidante enérgico, como el dicromato de potasio, en medio ácido sulfúrico con el ion plata (Ag+) como catalizador. En el caso de la volumetría, la determinación se realiza por valoración del exceso de dicromato no reducido, y la materia oxidable se calcula en términos de equivalente de oxígeno. En el caso de la colorimetría la determinación se realiza después de finalizada la digestión, pues se produce la reducción del Cr6+ del ion dicromato a Cr3+ (verde), que absorbe en la región de los 600 nm, donde el dicromato tiene absorción casi nula. Para DQOs en el rango 100-900 mg/L, se determina el incremento en Cr3+ a 600 nm, y valores más altos pueden ser obtenidos por dilución.46 Existen equipos que realizan la digestión y también la determinación de manera rápida y sencilla 32, pero se puede realizar la digestión en un bloque de calentamiento y luego ejecutar la lectura en un espectrofotómetro.
Determinación de ácidos grasos volátiles
Los AGV, principalmente el ácido acético, pueden estar contenidos en la biomasa sin pretratar, aunque también son subproductos de algunos pretratamientos, como en el pretratamiento por agua caliente presurizada. Aquí el ácido acético obtenido por la hidrólisis del grupo O-acetil contenido en la hemicelulosa, genera iones H+, que incrementan la eficacia del pretratamiento solubilizando prácticamente toda la hemicelulosa y parte de la lignina.48 En pretratamiento de cachaza con NaOH, 49 encontró como AGV mayoritarios los ácidos acético y butírico, mientras que en el pretratamiento termoalcalino de la cachaza con Ca(OH)2 realizado por López González 50, el ácido acético fue el único identificado. Su generación depende principalmente de la temperatura, el tiempo de pretratamiento y la carga de álcalis empleada en esos casos.
Existen diversos métodos para la determinación de AGV: destilación, espectrofotometría 51, GC 30,52, HPLC 53, siendo este último el más empleado en la evaluación del efecto de las técnicas de pretratamiento. También se puede realizar el análisis de ácidos orgánicos volátiles en la FL a través de lavaloración potenciométrica y la determinación de alcalinidad.47
La alcalinidad total es la capacidad del agua para neutralizar ácidos, debido a la presencia de sustancias como bicarbonato, carbonato e hidróxido, amoníaco, borato, fosfato, silicato y los aniones orgánicos que también pueden incluirse. Es aproximadamente equivalente a la concentración del anión bicarbonato para sustratos que tengan baja la concentración de AGV. Sin embargo, cuando la concentración de AGV se incrementa, estos son neutralizados por la alcalinidad al bicarbonato, y la alcalinidad total está compuesta por ambas, o sea, al bicarbonato y a los AGV. La muestra se valora con una solución de ácido normalizada hasta pH=5 y pH=4,4, de acuerdo con el método del Centro Federal Alemán de Investigación Agrícola (FAL), y para pH=5, pH=4,3, y pH=4,0, de acuerdo con Kapp.47 Estos puntos finales se utilizan en la determinación de la alcalinidad parcial (AP), debida a los bicarbonatos y la intermedia (AI), que indica la concentración de AGV.
Determinación de productos de degradación de azúcares
La FL también puede contener productos de degradación de carbohidratos, como HMF y furfural, así como otros componentes de interés, como ácidos orgánicos y alcoholes de azúcar. Los azúcares monoméricos se cuantifican mediante HPLC con detector de índice de refracción, así como los azúcares oligoméricos (después de convertirse a la forma monomérica mediante hidrólisis ácida) y sus productos de degradación.38
Producto de la degradación de pentosas y hexosas que conforman los azúcares, se forma el furfural y el HMF, que afectan negativamente los procesos de digestión anaerobia. Existen varios métodos para la determinación de ambos componentes: cromatografía de gases, HPLC y espectrofotometría. El más utilizado y más exacto es el de HPLC 7,54, pero la espectrofotometría constituye un método más sencillo, que conlleva equipos más simples y es mucho más rápido (tabla 4), y ha mostrado tener resultados comparables al HPLC-UV en cuanto a selectividad, linealidad y exactitud en el análisis realizado en sirope de maíz, realizado por De Andrade y colaboradores.55

Determinación de productos de solubilización de la lignina
Debido a la solubilización de la lignina durante los pretratamientos, pueden formarse derivados fenólicos, entre ellos los ácidos ferúlico (ácido 3-(4-hidroxi-3-metoxifenil)-2-propenoico), p-cumárico (ácido 3-(4-hidroxifenil)-2-propenoico), sirínguico (ácido 4-hidroxi-3,5-dimetoxibenzoico), vanílico (ácido 4-hidroxi-3-metoxibenzoico) y gálico (ácido 3,4,5-trihidroxibenzoico) 19, que tienen efectos inhibitorios sobre procesos de bioconversión, como la digestión anaerobia, por lo que deben ser cuantificados. El método más utilizado con este fin en la evaluación de los pretratamientos a biomasas lignocelulósicas es el de HPLC 19, aunque existen otras métodos más convencionales que resultan confiables y pueden utilizarse, como el de Folin-Cicolteau 57,58, basado en la reacción de los compuestos fenólicos con el reactivo de Folin-Cicolteau (ácido fosfomolibdotúngstico, de color amarillo) en medio básico, para originar, por reducción, una mezcla de óxidos de color azul, cuya absorbancia se mide a 765 nm.
Determinación cualitativa de cambios estructurales en la biomasa lignocelulósica
Es necesario destacar que el análisis composicional no es suficiente para investigar los efectos de un pretratamiento sobre la biomasa lignocelulósica. Es decir, no es suficiente saber la cantidad de lignina que tiene una biomasa, sino también dónde se encuentra, y la forma en que interactúa con la celulosa y la hemicelulosa. Su relocalización producto del pretratamiento es tan importante como su eliminación en la mejora de la hidrólisis de la biomasa, ya que puede influir en procesos posteriores de bioconversión.
Actualmente, la SEM, TEM y AFM, son ampliamente utilizadas para investigar las estructuras de materiales lignocelulósicos a escalas nanométricas que no se pueden deducir por otros análisis (figura 3). Con ellas se pueden observar las microfibrillas y las paredes celulares. SEM y TEM pueden proporcionar imágenes bidimensionales, mientras que una alta resolución con imágenes tridimensionales puede ser obtenida por AFM sin la necesidad de preparar la muestra, teñirla, deshidratarla o cubrirla con algún metal para poner de relieve las estructuras de la pared celular y sus componentes.59
Otro factor importante es la cristalinidad de la celulosa. A diferencia del almidón y la hemicelulosa, esta tiene una estructura que desempeña una función importante en la conversión biológica. Una evidencia de esto es la muy baja digestibilidad enzimática y microbiana de las fibras de algodón natural puro, en las que no está presente lignina ni hemicelulosa. La celulosa está formada en regiones con un orden molecular bajo (regiones amorfas o celulosa), regiones con un orden cristalino muy alto (celulosa cristalina), y una pequeña cantidad de la materia con un orden intermedio. Las regiones amorfas son capaces de adsorber el agua; además, su hidrólisis química, enzimática, y microbiana son más fáciles y más rápidas que para las zonas cristalinas. La celulosa amorfa se puede obtener de la celulosa cristalina por medio de los pretratamientos; sin embargo, en presencia de agua, la celulosa amorfa construida es termodinámicamente inestable y regresa en parte a su forma cristalina.6
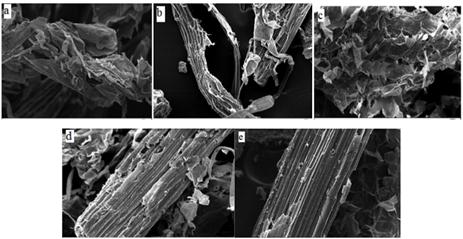 Fuente: Nosratpour y colaboradores 39
Fuente: Nosratpour y colaboradores 39
Fig. 3 Imágenes SEM (× 500) de (a) Pretratamiento con carbonato de sodio a 180 °C, (b) Pretratamiento con carbonato de sodio a 140 °C, (c) Pretratamiento con sulfito de sodio a 140 °C, (d) Pretratamiento con acetato de sodio a 180 °C, y (e) Bagazo sin pretratar
Se ha informado en varias ocasiones que la “reducción de la cristalinidad” de la celulosa resulta en mayores velocidades de bioconversión.59 Sin embargo, diversas investigaciones han demostrado que una mayor digestibilidad se puede obtener con mayor cristalinidad. En esos casos, otros factores (área superficial accesible, porosidad, contenido de lignina y hemicelulosa, y tamaño de partícula) fueron los de mayor influencia. La cristalinidad puede ser analizada por XRD, FTIR y RMN. FTIR es un método adecuado para propósitos comparativos, mientras que los valores del índice de cristalinidad son más precisos por los análisis de XRD y de RMN.6
Conclusiones
A partir del análisis de las técnicas empleadas en la evaluación del efecto de los pretratamientos, se concluye que: los métodos de análisis de carbohidratos estructurales y lignina de Weender y Van Soest continúan siendo utilizados para el análisis de biomasas lignocelulósicas pretratadas, pero se deben tener en cuenta varios aspectos para realizar los procedimientos; los métodos cromatográficos permiten identificar los monosacáridos presentes en la fracción líquida, pero los métodos colorimétricos como los de Dubois y Miller permiten un análisis simple y más rápido de las muestras; la potenciometría resulta una vía alternativa al HPLC muy atractiva para la determinación de AGV en muestras pretratadas; y es importante tener en cuenta que no solo basta con conocer la variación en la composición y eliminación de los componentes con el pretratamiento, también es necesario conocer su relocalización y cómo cambia su interacción con los demás componentes, a partir del empleo de técnicas como la microscopía electrónica de barrido y la microscopía de fuerza atómica.
Listado de abreviaturas


