










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Farmacia
versión On-line ISSN 1561-2988
Rev Cubana Farm vol.49 no.2 Ciudad de la Habana abr.-jun. 2015
ARTÍCULO ORIGINAL
Validación del método m―hidroxibifenilo para la cuantificación de ácido urónico en polisacáridos purificados de Streptococcus pneumoniae
Validation of the m-hydroxibiphenil method for the quantitation of uronic acid in purified Streptococcus pneumoniae polysaccharides
Lic. Marylín Pérez Calixto,I Dra. C. Yania Suárez Pérez,II MSc. Humberto González Rodríguez,I Téc. Lay Achong García,I Yohana Borrell CastilloI
I Instituto Finlay, Centro de Investigación‒Producción de Vacunas y Sueros. La Habana, Cuba.
II Departamento de Tecnología y Control de Medicamentos. Instituto de Farmacia y Alimentos (IFAL). Universidad de La Habana. La Habana, Cuba.
RESUMEN
Introducción: la complejidad y diversidad estructural de los polisacáridos capsulares de Streptococcus pneumoniae hace que la metodología analítica necesaria para la caracterización y control de calidad sea engorrosa. El método colorimétrico m‒hidroxibifenil desarrollado por Blumenkrantz y Asboe‒Hansen en el año 1973, permite contrarrestar las desventajas del método propuesto en la Farmacopea Europea 6ta Edición, 2011.
Objetivo: validar el método m‒hidroxibifenilo para cuantificar ácido urónico en polisacáridos purificados de streptococcus pneumoniae.
Métodos: se determinó el tiempo de vigencia de la solución de tetraborato de sodio responsable de generar la respuesta analítica y se seleccionó una metodología para la cuantificación de ácido urónico por el método del m‒hidroxibifenilo se emplea una solución estándar de ácido galacturónico a 1 mg/mL. Se realizó una evaluación de los parámetros de validación: linealidad, exactitud, precisión, rango y especificidad, según exigencias actuales. Además se comparó con el método del carbazol descrito en la Farmacopea Europea 6ta Edición, 2011 para la cuantificación de ácido urónico.
Resultados: se demostró que el tiempo de vida útil de la solución de tetraborato de sodio fue de 24 h. El método del m‒hidroxibifenilo fue específico, lineal, exacto y preciso en el rango de 2‒20 μg/mL porque se cumplieron satisfactoriamente los criterios de aceptación establecidos para cada uno de ellos. La comparación del método propuesto con el método normalizado reveló que no existieron diferencias estadísticamente significativas entre ambos.
Conclusión: el método colorimétrico del m‒hidroxibifenilo resultó válido para el control de calidad de las muestras de polisacárido purificado de Streptococcus pneumoniae, dando resultados comparables con el método recomendado en la Farmacopea Europea, 2011.
Palabras clave: validación, colorimetría, m‒hidroxibifenilo, ácido galacturónico, Streptococcus pneumoniae.
ABSTRACT
Introduction: complexity and diversity in the structure of Streptococcus pneumoniae capsular makes the analytical methodology for characterization and quality control a troublesome aspect. The colorimetric m/hydroxibiphenil method devised by Blumenklrantz and Asboe-Hansen in 1973 allows reducing the disadvantages of the suggested method in the European Pharmacopea 6th edition, 2011.
Objective: to validate the m-hydroxybiphenil method for quantitation of uronic acid in purified Streptococcus pneumonia polysaccharides.
Methods: the length of validity of a sodium tetraborate solution, responsible for generating the analytical response, was estimated, and additionally, a methodology for quantitation of uronic acid by the m-hydroxybiphenil method was selected in which a standard galacturonic acid solution at 1mg/ml was used. The validation parameters were evaluated as follows: linearity, accuracy, precision, range and specificity, according to the present requirements. This method was compared with the carbazol method described in the European Pharmacopea 6th edition, 2011 for quantitation of uronic acid.
Results: it was demonstrated that the useful lifetime of the sodium tetraborate solution was 24 hours. The m-hydroxybiphenil method was specific, linear, accurate and precise in the range of 2 to 20 ?g/mL because the set acceptance criteria were satisfactorily complied with for each of them. The comparison of the suggested method with the standardized method yielded that no statistically significant differences exist between them.
Conclusions: the colorimetric m-hydroxybiphenil method proved to be valid for the quality control of purified Streptococcus pneumonia polysaccharide samples and the results are comparable to those of the recommended method in the European Pharmacopea, 2011.
Keywords: validation, colorimetry, m-hydroxybiphenil, galacturonic acid, Streptococcus pneumonia.
INTRODUCCIÓN
Streptococcus pneumoniae es uno de los patógenos más importantes responsable de una gran variedad de infecciones invasivas, entre las que se encuentran la sepsis, la meningitis y la neumonía, muy frecuentes en niños, adultos mayores de 60 años y pacientes inmunocomprometidos.1,2 La incidencia de la neumonía en los últimos años se ha incrementado notablemente y constituye un problema de salud de primera magnitud, ya que es una de las principales causas de muerte y de ingreso hospitalario. La Organización Mundial de la Salud (OMS) estima que al menos un millón de niños menores de 5 años mueren cada año de neumonía por Streptococcus pneumoniae.3
El Instituto Finlay y el Centro de Química Biomolecular se dieron a la tarea de obtener un candidato vacunal que cubra un amplio espectro de los serotipos de mayor incidencia en Cuba. Una vez lograda la vacuna, el Sistema de Salud cubano podrá contar con una nueva opción en el Esquema Nacional de Inmunización, la que se exportará a países pobres.
Es indudable el rigor que debe primar en el establecimiento de la metodología de evaluación de las materias primas y del producto final. Desde el punto de vista regulador, el diseño de una metodología analítica debe estar sustentado en una adecuada selección de los métodos, en la ausencia de redundancia de resultados y en la demostración de que una determinada estructura bien caracterizada garantiza una función específica.4
La complejidad y diversidad estructural de los polisacáridos capsulares de Streptococcus pneumoniae hace que la metodología analítica necesaria para su caracterización y control de calidad sea engorrosa. De ahí la importancia de validar los métodos que se empleen, para asegurar la confiabilidad de los resultados y cumplir con los requerimientos de las Buenas Prácticas de Producción para la Fabricación de Productos Farmacéuticos y en especial de las vacunas para uso humano, ya que la validación constituye la confirmación por examen y evidencias objetivas de que se han cumplido los requisitos particulares para su utilización.5
La Farmacopea Europea 6ta Edición, 20116 recomienda el empleo del método colorimétrico del carbazol para la determinación del contenido de ácido urónico en los polisacáridos purificados de Streptococcus pneumoniae. Este método tiene entre sus desventajas el empleo de una mayor cantidad de muestra, así como un elevado tiempo de procesamiento. Sin embargo el método colorimétrico m‒hidroxibifenil desarrollado por Blumenkrantz y Asboe—Hansen en el año 1973,7 permite una disminución en el consumo de muestras, además de ser un método más rápido, sensible y que emplea una solución colorante más estable.
Basado en este criterio se propuso en este trabajo validar el método m‒hidroxibifenilo para la cuantificación de ácido urónico en polisacáridos purificados de streptococcus pneumoniae.
MÉTODOS
DETERMINACIÓN DE LA CONCENTRACIÓN DE ÁCIDO URÓNICO EMPLEANDO EL MÉTODO DEL M-HIDROXIBIFENIL. DESCRIPCIÓN DEL MÉTODO
Preparación de la curva de calibración
Se descongeló un vial que contiene 1 mL de la solución de ácido galacturónico a 1 mg/mL [solución de referencia (SR): lote‒S6206623‒Merck]. Se adicionaron alícuotas de 10, 20, 40, 80 y 100 μL de la SR a tubos de ensayo, y se completó a 2 mL con agua purificada. Se tomaron 400 μL por triplicado de cada una de las soluciones preparadas anteriormente para obtener 2, 4, 8, 16 y 20 μg/mL de ácido galacturónico en cada punto de la curva.
Preparación de las muestras
Se determinó el porciento de humedad residual al polisacárido seco en el analizador de humedad SARTORIUS MA 100. En una navecilla se colocó aproximadamente 120 mg de la muestra a analizar, la misma se sometió a un proceso de secado a una temperatura de 60 °C durante una hora. El porciento de humedad para los polisacáridos purificados se obtuvo directamente en la balanza. El mismo debe ser menor que el 10 %.
El polisacárido purificado seco (una vez determinado el porciento de humedad), se disolvió en agua purificada, hasta una concentración de 5 mg/mL. A la solución de polisacárido purificado serotipo 1 se le realizó una dilución 1/100 y para el caso de la solución de polisacárido purificado serotipo 5 se le realizó una dilución 1/50.
Procedimiento analítico
Las disoluciones preparadas para la curva de calibración, el blanco (400 μL de agua purificada) y las muestras, se introdujeron en un baño de agua fría. Posteriormente se le adicionó a cada tubo de ensayo 2,4 mL de una solución de tetraborato de sodio decahidratado 0,125 mol/L y se agitó vigorosamente en vortex. Seguidamente los tubos se colocaron en un baño de agua a 100 °C durante cinco minutos, transcurrido este tiempo, se extrajeron los tubos y se detuvo la reacción dejándolo enfriar introduciéndolos en un baño de agua helada durante algunos minutos. A continuación se añadieron 40 μL de la solución m‒hidroxifenilfenol en hidróxido de sodio a todos los tubos excepto al blanco al que se le añadió 40 μL de una solución de NaOH 0,5 %. Se agitó concienzudamente y se leyó inmediatamente la absorbancia en el espectrofotómetro ULTROSPEC 2100 a una longitud de onda 520 nm contra blanco de compensación.
ESTIMACIÓN DEL TIEMPO DE VIDA ÚTIL DE LA SOLUCIÓN TETRABORATO DE SODIO
Para determinar el tiempo de vida útil de la solución de tetraborato de sodio en ácido sulfúrico 0,125 mol/L; se prepararon 3 soluciones y el ensayo se realizó por triplicado en tres días diferentes. Se compararon los resultados obtenidos a través de un análisis de varianza empleando el programa STATGRAPHICS plus 5.1, opción “Comparación”, “Análisis de varianza”, “ANOVA simple”. Además se graficaron los valores medios para cada solución en cada día, empleando gráficos de línea.
El método utilizado en el ensayo se validó de acuerdo con los criterios establecidos.5 Los parámetros evaluados fueron los siguientes:
Linealidad: se realizó el ensayo por triplicado en 3 días diferentes de todos los puntos de la curva de calibración (2, 4, 8, 16 y 20 μg/mL). Se determinó la ecuación de la recta de regresión, se calculó el coeficiente de correlación lineal (r), coeficiente de determinación (r2) y el coeficiente de variación (CV) de los factores respuesta analítica (absorbancia/concentración) de cada punto de la curva.
Exactitud: se empleó el método de dilución de la solución de referencia. A partir de la SR de ácido galacturónico 1 mg/mL, se prepararon diluciones para obtener 2, 8 y 20 μg/mL de concentración teórica respectivamente. El ensayo se realizó en cuatro días diferentes. Se determinó el recobrado medio, la desviación estándar de los porciento de recobro (R) y el coeficiente de variación total (CV). También se realizó la prueba de Cochran para determinar influencia de la concentración en la variabilidad de los resultados, así como la prueba de t de Student para demostrar la presencia o no de diferencias significativas del porciento de recobrado y con el 100 %.
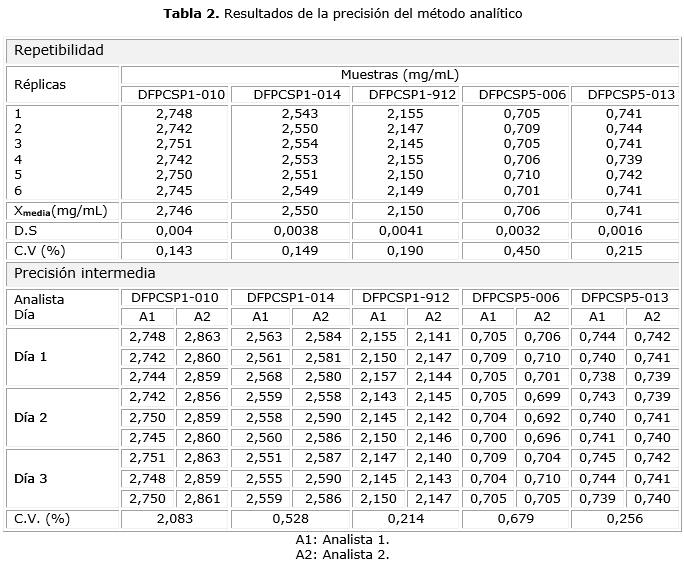
Precisión: se determinó la repetibilidad por el análisis sextuplicado de la misma muestra y la precisión intermedia a través del análisis por triplicado de muestras analizadas por dos analistas en tres días diferentes. En ambos casos, se calculó la media, la desviación estándar y el coeficiente de variación.
Especificidad: para el análisis de la especificidad se llevaron a cabo dos variantes:
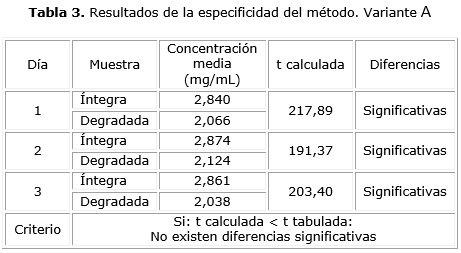
Variante A: se realizó la evaluación de la influencia de los posibles productos de degradación. Para ello la muestra de polisacárido purificado (lote DFPCSP1—001) se sometió a un proceso de degradación introduciéndola en un horno a una temperatura de 100 °C durante 2 h. La degradación de la muestra se corroboró mediante un análisis por resonancia magnética nuclear protónica (RMN1 H) y por la determinación de la constante de distribución (KD). Se compararon las concentraciones medias de ácido urónico obtenidas en las muestras íntegras y las degradadas por triplicado durante tres días consecutivos empleando una prueba t de Student.
Caracterización por RMN1H de los productos de degradación: la caracterización de los polisacáridos capsulares (PsC) y de todos los derivados obtenidos, se realizó por RMN1 H a 325 K con 128 acumulaciones (Brucker AC-250F, 250 MHz). Para ello se disolvieron 5 mg de cada muestra en 0,6 mL de agua deuterada (D2O), posteriormente se liofilizaron a sequedad. Se reconstituyó el volumen en 0,6 mL de D2O y los espectros se realizaron a una temperatura de 52 ºC. Una vez procesados los espectros por el programa MestRe—C, se pudieron asignar y evaluar las señales características de los polisacáridos, definiendo identidad estructural y la conservación de la estructura.
Determinación de la constante de distribución (KD): en la determinación de la KD se empleó la Cromatografía de Exclusión Molecular en un equipo de Cromatografía Líquida de Alta Resolución (HPLC) (KNAUER Smartline 5000) con columna TSK 5000 PW. Se utilizó como fase móvil una disolución de NaCl 0,9 % y como detector IR (Knauer, Smartline, 2 300). Las muestras disueltas en agua se inyectaron a una concentración de 5 mg/mL. Una vez que se obtuvo el perfil cromatográfico, se determinó el tiempo de retención (tr) en el punto máximo del pico y se calculó la KD de cada muestra.
KD = (Vr‒Vo) ̸ (Vt‒Vo)
Donde:
Vo: volumen extra particular
Vr: volumen de retención
Vt: volumen total de la columna
El valor de KD se calculó según la fórmula:
% discrepancia= (C muestra cargada — C muestra sin cargar) ̸ C muestra sin cargar
Donde:
C muestra cargada: Concentración de la muestra con glucosa.
C muestra sin cargar: Concentración de la muestra sin glucosa.
Variante B: se evaluó la capacidad del método para identificar solo el analito de interés en presencia de otras sustancias. Se preparó una solución de glucosa a una concentración de 3 mg/mL. Se adicionaron 20, 40 y 80 μL de la misma a las muestras (lotes DFPCSP1-014, DFPCSP1-912, DFPCSP5-006). El ensayo se realizó por triplicado durante tres días diferentes. Se determinó el porciento de discrepancia.

El porciento de discrepancia obtenido debe ser menor que el coeficiente de variación del método calculado como el valor histórico obtenido en la determinaciones efectuadas para esta técnica.
COMPARACIÓN DE LOS MÉTODOS
Se ensayaron muestras por triplicado durante 5 días consecutivos, con el empleo de ambos métodos (método propuesto en este trabajo y método recomendado en la Farmacopea Europea).6 Se determinaron las concentraciones medias y los CV de los resultados de cada muestra. Para evaluar la influencia de los factores días y métodos se realizó una comparación entre las varianzas mediante un ANOVA simple empleando el programa STAGRAPHICS Plus 5.1.
RESULTADOS
ESTIMACIÓN DEL TIEMPO DE VIDA ÚTIL DE LA SOLUCIÓN TETRABORATO DE SODIO
Los resultados obtenidos se muestran en la figura. Como se observa, la solución de tetraborato de sodio a 0,125 mol/L puede ser empleada durante un tiempo no superior a 24 h. Se comprobó que existieron diferencias estadísticamente significativas entre los resultados obtenidos en días diferentes, con un valor de F calculado (79,512) > F tabulado (5,053).

RESULTADOS DE LA VALIDACIÓN DEL MÉTODO ANALÍTICO
A continuación se presentan los resultados obtenidos en la validación del método de m‒hidroxibifenilo empleado para la cuantificación de la concentración de ácido urónico en muestras de polisacáridos purificados de Streptococcus pneumoniae.
Los gráficos de las tres curvas de patrones ensayadas para determinar la linealidad del método fueron muy similares para los tres días estudiados. El análisis de regresión permitió comprobar que los valores de todos los parámetros de las curvas de calibración fueron adecuados, confirmando la linealidad de las tres experiencias realizadas en días diferentes (tabla 1).
Los resultados de la evaluación de la exactitud demostraron que en los niveles de concentración evaluados los porcientos de recobro se encontraron en el rango establecido (97—103 %) para métodos químicos y espectrofotométricos, así como el recobrado medio (99,89 %), el cual no difiere estadísticamente del 100 % (t experimental= 1,46 > t tabulada= 2,16). El coeficiente de variación total (CV= 1,54 %) fue inferior al 3,0 %. El cumplimiento de todos los criterios establecidos al procesar los resultados de la curva de recuperación correspondiente, demostró la correlación entre la concentración teórica y experimental (tabla 1). La evaluación de la influencia de la concentración de analito en la varianza de los resultados, a través de la Prueba G de Cochran, demostró que las varianzas de los tres niveles de concentración evaluados fueron equivalentes, ya que G experimental (0,4300) ˂ G tabulada (0,8709).
En la evaluación de la repetibilidad y de la precisión intermedia, los CV obtenidos fueros muy inferiores al límite establecido de 3,0 %, por lo que el método cumplió con los criterios de aceptación vigentes5 (tabla 2).
El análisis de varianza para determinar el efecto del día y del analista en los resultados de la concentración de ácido urónico para cada muestra, dio resultados satisfactorios ya que no se obtuvieron diferencias estadísticamente significativas para una p≤ 0,05 entre los resultados experimentales obtenidos por los dos analistas; independientemente del día en que se efectuó el análisis; ni entre los resultados obtenidos en cada día, independientemente del analista que aplicó el método en estudio.
Al aplicar la variante A para el ensayo de especificidad, las concentraciones medias resultantes de la evaluación de las muestras íntegras y degradadas, fueron diferentes, ya que en todos los casos las t calculadas fueron superiores a la t tabulada (1,860) (tabla 3), por lo que las diferencias encontradas desde el punto de vista estadístico fueron significativas para una p≤ 0,05; lo cual demostró la capacidad del método de discriminar la respuesta analítica.
Los resultados obtenidos para los porcientos de discrepancia con la variante B cuando se le adicionaron 20 y 40 μL de glucosa respectivamente, estuvieron por debajo del coeficiente de variabilidad promedio del método que fue de 4,77 %. Sin embargo, para las muestras cargadas con 80 μL los porcientos de discrepancias fueron superiores al coeficiente de variabilidad promedio (tabla 4), por lo que la técnica no fue específica cuando los volúmenes de glucosa son de 80 μL.
COMPARACIÓN DE LOS MÉTODOS ANALÍTICOS
Los resultados obtenidos durante la comparación de ambos métodos se obtuvieron valores similares de concentración de ácido urónico (tabla 5). No existieron diferencias significativas cuando se compararon las varianzas por el ANOVA simple entre los resultados obtenidos por ambos métodos, en los cinco días de estudio.
DISCUSIÓN
ESTIMACIÓN DEL TIEMPO DE VIDA ÚTIL DE LA SOLUCIÓN TETRABORATO DE SODIO
Se consideró en este estudio que muchas de las soluciones usadas en el laboratorio, con frecuencia, son inestables a la temperatura de trabajo o se contaminan fácilmente con lo que se originan alteraciones en sus propiedades físicas y químicas, por lo que resulta de gran importancia definir el período de validez o tiempo de vida útil de las mismas, una vez que son preparadas. Se comprobó que existieron diferencias estadísticamente significativas entre los resultados obtenidos en días diferentes, lo cual determinó el período máximo de validez de la solución de tetraborato de sodio a 0,125 mol/L igual a 24 h, al transcurrir este tiempo comienzan a existir fluctuaciones en los valores de concentración de ácido urónico que pudieran provocar errores en la determinación (Fig.). No obstante, aun se considera que es buena la estabilidad alcanzada en la solución de tetraborato de sodio a 0,125 mol/L.
RESULTADOS DE LA VALIDACIÓN DEL MÉTODO ANALÍTICO
El método fue lineal ya que los coeficientes de regresión y determinación durante los tres días de estudio, fueron superiores a los establecidos5 y por otra parte los factores de respuesta tuvieron muy poca variación, lo cual se reflejó por los valores promedios de CVf, los cuales fueron inferiores al 5 % establecido como límite. Estos resultados avalan la proporcionalidad existente entre la respuesta analítica y la concentración del analito en el rango analizado de 2‒20 μg/mL. En todos los casos el intercepto no difiere significativamente de cero, ya que las t calculadas ˂ t tabulada. Las pendientes dieron valores de t experimentales altos y la probabilidad fue inferior a 0,05. Un comportamiento similar se presentó para la exactitud. Además del cumplimiento satisfactorio de los parámetros establecidos para evaluar la curva de recuperación por regresión lineal, se comprobó que no influyó el factor de concentración en la variabilidad de la respuesta medida. El test de Student corroboró la exactitud, ya que la t calculada ˂ t tabulada, por lo que no existieron diferencias estadísticamente significativas entre el recobrado medio y el 100 % para una p≤ 0,05. De este modo se puede afirmar que la técnica no se afectó por errores sistemáticos de forma significativa, por lo que se pueden obtener valores experimentales muy próximos al valor verdadero, para el análisis del contenido del analito en el nuevo método desarrollado, en los tres niveles de concentración evaluados. El conjunto de estos resultados confirmó la exactitud del método propuesto para el control de calidad de polisacárido purificado de Streptococcus pneumoniae, y se demuestra que el método en estudio no presenta errores por exceso ni por defecto, que provoquen sesgo de los resultados experimentales respecto a los valores reales.
El método fue suficientemente repetible independientemente de la muestra analizada. Se obtuvieron resultados experimentales con muy baja variabilidad al realizar análisis repetidos por el mismo analista, el mismo laboratorio y el mismo día. Los factores días y analistas no contribuyeron significativamente en la respuesta analítica, por lo que también se consideró adecuado desde el punto de vista de la precisión intermedia. Por esta razón, se consideró mínima la contribución de los errores aleatorios.
Uno de los parámetros más importantes es la especificidad, dado por el impacto que puedan tener los restantes componentes de la matriz que eventualmente actúen como potenciales interferencias. En este caso se consideró relevante el efecto de la degradación térmica (Variante A)y de la presencia de glucosa (Variante B). El método es capaz de discriminar entre analito y productos de degradación y entre analito y glucosa siempre que esta no se encuentre a concentraciones superiores a 40 μL. No obstante, se detectó un cambio de color inmediatamente después de ser extraídas las muestras de la etapa de calentamiento, revelando la presencia de sacáridos indeseados.
COMPARACIÓN DE LOS METODOS ANALÍTICOS
El método del m‒hidroxibifenil (Método 2), previamente validado para su aplicación a la cuantificación de ácido urónico en muestras de polisacárido purificado de Streptococcus pneumoniae, brinda resultados comparables al Método 1 de referencia (método de carbazol), reportado en la Farmacopea Europea 6ta Edición,6 resultado de gran importancia pues posibilita su aplicación con el objetivo previsto. El empleo del método colorimétrico del carbazol para la determinación del contenido de ácido urónico en los polisacáridos purificados de Streptococcus pneumoniae tiene entre sus desventajas el empleo de una mayor cantidad de muestra, así como un elevado tiempo de procesamiento. Sin embargo, el método colorimétrico m‒hidroxibifenil desarrollado por Blumenkrantz y Asboe‒Hansen en el año 1973,7 permite disminuir el consumo de muestras, además de ser un método más rápido, sensible y que utiliza una solución colorante relativamente más estable, de ahí la importancia de este trabajo. El método colorimétrico del m‒hidroxibifenilo resultó válido para el control de calidad de las muestras de polisacárido purificado de Streptococcus pneumoniae, con resultados comparables con el método recomendado en la Farmacopea Europea, 2011.
REFERENCIAS BIBLIOGRÁFICAS
1. Fedson DS. The clinical effectiveness of pneumococcal vaccination: a brief review. Vaccine. 1999;17(Suppl 1):85-90.
2. Bogaert D, de Groot R, Hermans PW. Streptococcus pneumoniae colonisation: the key to pneumococcal disease. Lancet Infect. Dis. 2004;4(3):144-54.
3. Schuchat A, Hilger T, Zell E. Active bacterial core surveillance of the emerging infection program network. Emerg. Infec. Dis. 2001;(7):1-8.
4. Centro Estatal de Control de Medicamentos. Regulación No. 37. Buenas Prácticas de Laboratorio. La Habana: CECMED; 2012:33-37.
5. Centro Estatal de Control de Medicamentos. Regulación No. 41. Validación de Métodos Analíticos. La Habana: CECMED; 2007:8-18.
6. Farmacopea Europea. Técnicas Analíticas para Vacunas Polisacarídica. 6ta Edición; 2011.
7. Blumenkrantz y Asboe‒Hansen. New method for quantitative determination of uronic acids. Analytical Biochemestry. 1973;(54):484-9.
Recibido: 24 de marzo de 2014.
Aprobado: 11 de septiembre de 2014.
Marylín Pérez Calixto. Instituto de Farmacia y Alimentos (IFAL), Ave 23 No. 21425 e/ 214 y 222. La Coronela, La Lisa. Ciudad Habana. Teléfonos: 2085176.
Correo electrónico: eneidasanchez@infomed.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}