










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Las miopatías inflamatorias idiopáticas (MII) son un grupo heterogéneo de enfermedades sistémicas y autoinmunes que afectan al músculo esquelético. Estas se caracterizan por debilidad proximal e inflamación del músculo esquelético, elevación de las enzimas musculares, patrón miopático en electromiografía, infiltrado inflamatorio en la biopsia del músculo, y anticuerpos asociados o específicos (Ac) de la enfermedad. Se reconocen diferentes variantes clínicas, la polimiositis (PM) y la dermatomiositis (DM) si tiene lesiones cutáneas, la miositis por cuerpos de inclusión (MCI), la miopatía asociada al cáncer, miositis necrotizante inmunomediada y los síndromes de superposición (SS), los que provocan alteraciones a nivel de otros órganos de la economía por lo que se consideran enfermedades sistémicas. Son enfermedades raras por su baja incidencia 11/1000000/año y prevalencia 14/100000. Con excepción de la dermatomiositis juvenil, las MII afectan mayormente a los adultos.1,2,3,4
En 1990 se descubrieron anticuerpos específicos como Mi2, la cual es un complejo de 8 proteínas asociada a lesiones cutáneas por lo que se encuentran presente en los pacientes con dermatomiositis, antipartículas de reconocimientos de señales (anti-SRP,) antihistidi (anti Jo-1), antitreonil (anti PL-7), antialanil (anti-PL-12), anti(glicil) antiEJ, antiisoleucil (anti OJ) y antiasparginil (anti-KS). El anticuerpo anti-histidil-ARNt es el más frecuente anticuerpo diferente al Anti-Jo-1 y su presencia confiere peor pronóstico. Los anticuerpos que incluyen anti-Ro, anti-La, anti-PM-Scl, anti-ribonucleoproteínas nucleares pequeñas (snRNPs) anti-Mas, anti-hPMS1 y el anticuerpo PM-Scl se asocia con un síndrome de superposición ligado a esclerosis sistémica con manifestaciones cutáneas mínimas y PM o DMPm/Scl.5,6
Los anticuerpos antisintetasa (anti-aminoacil-ARNtsintetasa), se encuentran en 40 % y se relacionan con el síndrome antisintetasa cuando este se caracteriza por presentar afección muscular, artritis no erosiva, fenómeno de Raynaud, “manos de mecánico” (fisuras laterales a nivel de los dedos de las manos), fiebre e importante afectación pulmonar intersticial que puede llevar a fibrosis del pulmón con la presencia de un anticuerpos antisintetasa (anti-aminoacil-ARNtsintetasa).6,7,8
El anticuerpo anti Mi-2 es especifico de miositis y se asocia a dermatomiositis tanto juvenil como del adulto, por estar relacionado con sus lesiones cutáneas, con poco riesgo de enfermedad pulmonar intersticial, y pronóstico bueno, con lesiones cutáneas persistentes, la debilidad muscular es leve y su prevalencia está en función de latitud geográfica y se relaciona con la incidencia de los rayos del sol.9,10
El anticuerpo melanoma differentiation-associated gene 5 (anti-MDA5) en la DM amiopática, el anticuerpo contra el factor intermediario de transcripción de proteínas (anti-TIF1) y el anticuerpo contra la proteína de matriz nuclear 2 (anti-NPX2) se asocian con la presencia de neoplasia en pacientes adultos con DM.10,11,12
Se describen anticuerpos antipartículas de reconocimientos de señales (anti-SRP) y anta 3-hydroxi--metil-glutaril-CoA reductasa (anti- HMG-Co A), el hallazgo de este anticuerpo apoya el diagnóstico de miositis inducida por estatinas. La presencia de estos dos anticuerpos (anti SRP y antiHMG-Co A) se asocian a miositis necrotizante inmunomediada (MNI).13,14,15
Las miopatías inflamatorias idiopáticas se caracterizan por la producción de auto anticuerpos específicos, marcadores muy útiles para el diagnóstico clínico, la clasificación y la predicción de su pronóstico, su presencia tiene diferentes implicaciones en función de la edad, raza y factores ambientales. Por lo antes expuesto, se realizó esta investigación con el objetivo de identificar las características clínicas, e inmunológicas y daño de órganos en los pacientes con miopatías inflamatorias idiopáticas.
Métodos
Se realizó estudio observacional, de corte transversal en el Hospital Clínico Quirúrgico “Hermanos Ameijeiras” en el periodo comprendido de enero de 2016 a enero de 2017.
Para la obtención de los datos se utilizó la historia clínica del paciente.
El universo fueron los pacientes mayores de 20 años atendidos en consulta protocolizada de miopatías inflamatorias idiopáticas del adulto que cumplieron los criterios del Colegio Americano de Reumatología1 (ACR) para diagnóstico de MII.
Dentro de los criterios de exclusión estuvieron los pacientes con enfermedad neurológica central o periférica y con infecciones del músculo estriado.
La muestra quedó conformada por 52 pacientes atendidos en el periodo de estudio.
Los pacientes se identificaron según el tipo de miopatía diagnosticada (dermatomiositis, polimiositis), variables sociodemográficas (edad, sexo, color de piel), tiempo de evolución de la enfermedad, estado de la enfermedad (actividad e inactividad o remisión, según la escala análoga del Medical Council Research) y antecedentes patológicos personales (hipertensión arterial y diabetes mellitus tipo 2).
Se identificaron manifestaciones clínicas de compromiso sistémico y la presencia o no de anticuerpos según dermatomiositis o polimiositis: anticuerpos específicos y asociados a miositis.
Características clínicas
La neumopatía intersticial se diagnosticó por clínica compatible (disnea de esfuerzo de reciente aparición, tos seca y fiebre sin otra causa), tomografía axial computarizada pulmonar de alta resolución y pruebas de función respiratoria.
La hipertensión pulmonar se definió por ecocardiograma (PSAP estimada ≥ 40 mmHg).
Las neoplasias demostradas por biopsia:
Osteoporosis, densidad ósea con un t score menor de 2,5 %
Daño renal cuando aparecen proteinurias secundarias a elevación de los niveles de CPK que provocan mioglobinuria.
Fenómeno de Raynaud: Cambio de coloración de las manos, cianosis seguida de palidez y después rubicundez.
Artritis: aumento de volumen de la articulación, rubor, dolor e impotencia funcional.
Disfagia: dificultad para tragar alimento líquidos y sólidos por debilidad de los músculos faríngeo por la actividad de la enfermedad.
Anticuerpos
Los que se informaron como presentes al colocar una muestra de sangre sobre la tira de anticuerpos detectados por el ensayo multiplex LA (line IMMUNE ASSYS, Human GBD).
Anticuerpos específicos de miositis y Ac asociados a miositis. Tipo de Anticuerpos detectados por el ensayo multiplex LA (line IMMUNE ASSYS, Human GBD), Wiesbaden, Alemania para determinar Ac como Hep-2, DNA ds, Nuc, Hist, SmD1, PCNA PO (RPP) Ro60, Ro52, La, Cen B, Scl70, U1RNP y AMAM2, anti Jo1, anti Mi 2, anti PL 7 y anti PL 12.
Dentro de los anticuerpos específicos tenemos: Anti Jo-1, Anti Mi-2, Anti SRP, Anti PL-12, Anti PL-7.
Dentro de los anticuerpos asociados: Ro60, Ro52, Ku, PMScl
Se confeccionó una base de datos en formato Excel de la Microsoft Office versión XP, la que fue posteriormente exportada al sistema SPSS versión 20.0 para su análisis. Se utilizaron estadígrafos descriptivos como la media aritmética y la desviación estándar para todas las variables cuantitativas continúas que se analizaron. Se confeccionaron histogramas para elaborar las escalas de clasificación.
Para las variables cualitativas se calcularon los porcentajes de cada grupo.
Se utilizó Chi-cuadrado de Pearson (Estadístico exacto de Fisher). (Nivel de significación del 95 % (α=0,05) para relacionar la presencia de anticuerpos y el tipo de miopatía, así como la presencia de manifestaciones clínicas de MII.
Consideraciones bioéticas
El estudio se realizó de acuerdo con lo establecido en la Declaración de Helsinki, modificación de Hong Kong sobre las investigaciones en seres humanos.
Resultados
La edad media de aparición fue de 42,8 ± 13,2 meses (42,6 ± 12,8 para DM y 43,2 ± 14,1 para PM). El tiempo medio de demora al diagnóstico fue de 8,8 ± 7,0 meses (9,0 ± 8,4 para DM y 8,7 ± 6,8 para PM). El tiempo medio de evolución de la enfermedad fue de 7,5 ± 7,1 años (6,2 ± 7,0 para DM y 10,2 ± 8,9 para PM). Un total de 42 (80,8 %) enfermos estaban en remisión en el momento del estudio (28/84,8 % con DM y 14/73,7 % con PM). Por otra parte, 15 (28,8 %) tenía HTA como antecedente patológico personal, con DM eran 12 (36,4 %) y con PM eran 3 (15,8 %), seguido de enfermedad tiroidea para un total de 6 (11,5 %) y diabetes mellitus tipo II con 4 casos (7,7 %) (tabla 1).
Tabla 1 Distribución de los pacientes según variables sociodemográficas, tiempo de evolución y estado de la enfermedad y antecedentes patológicos personales
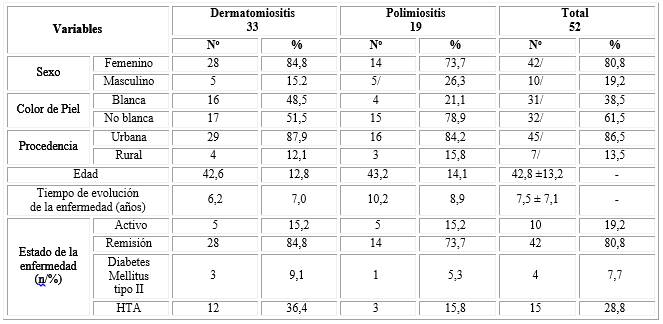
±: Desviación estándar
APP: antecedente patológico personal
HTA: hipertensión arterial.
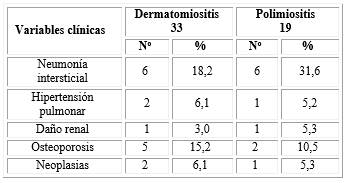
La tabla 2 muestra que 12 pacientes presentaron neumonía intersticial (23,1 %) (6, (18,2 %) con DM y 6, (31,6 %) con PM). La osteoporosis se encontró en 5 pacientes con dermatomiositis (15,2 %) y la hipertensión pulmonar en 2 (6,1 %). Solo un paciente con polimiositis (5,2 %), la neoplasia se encontró en 2 pacientes con dermatomiositis (6,1 %) y solo 1 paciente en la polimiositis (5,3 %). El daño renal se encontró en 2 (3 %) pacientes, uno con polimiositis (5,3 %) y otro con dermatomiositis.
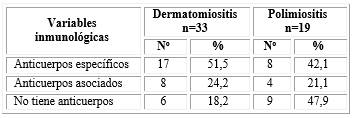
Los anticuerpos de miositis estuvieron en 25 pacientes (48,1 %) se presentaron Ac específicos, de ellos 17/51,5 % tenía DM y 8/42,1 % PM. Los anticuerpos asociados se encontraron en 8/24,2 % de DM y 4/21,1 % de PM, y no presentan ningún anticuerpo 6/18,2 % de DM y 9/47,9 % de PM (tabla 3).
Los anticuerpos que más se encontraron en estos casos fueron los anticuerpos específicos de miositis, de ellos el Anti-Jo 1 en 14 pacientes, con DM 7 y con PM otros 7 (77,7 %), seguido por el Anti-Mi-2 con 8 casos, siendo todos los enfermos de DM y presentando este último anticuerpo significación estadística, mientras que los asociados que más se encontraron fueron el Ro 52 en 10, de estos con DM 6 (35.2%) y con PM 4 (44,4 %) y el Ro60 con 6, de ellos 3 (17,6 %) con DM y otros 3 (33,3 %) con PM (tabla 4).
Tabla 4 Anticuerpos específicos y asociados a miopatías inflamatorias
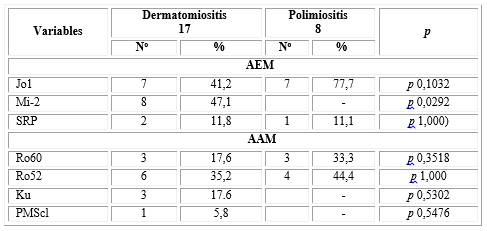
*Chi-cuadrado de Pearson (Estadístico exacto de Fisher)
Las manifestaciones clínicas con mayor presencia de Ac específicos y asociados fueron neumonía intersticial con 10 (40 %) y 2 (16,7 %) respectivamente, el fenómeno de Raynaud con 11 (44 %) y 4 (33,3 %), el eritema heliotropo con 15 (60 %) y 11 (91,7 %), la pápula de Gottron con 13 (52 %) y 11 (91,7 %), el signo de Gottron con 12 (48 %) y 10 (83,3 %), el eritema facial con 15 (60 %) y 11 (91,7 %) y en zonas fotoexpuestas con 12 (48 %) y 11 (91,7 %) y el signo de la pistolera con 4 (16 %) y 9 (75 %), resultados todos con significación estadística (tabla 5).
Tabla 5 Presencia de anticuerpos y manifestación clínica de las miopatías inflamatorias idiopáticas

*Chi-cuadrado de Pearson (Estadístico exacto de Fisher).
Según las manifestaciones clínicas el Anti- Jo 1 se observó en 14 (100 %) casos con artritis, en 9 (64,3 %) con neumonía intersticial y en 8 (57,1 %) con eritema facial y en zonas foto expuestas. El Mi-2 en 8 (100 %) pacientes con DM, artritis y lesiones cutáneas, cada una de ellas. El Anti- SRP se encontró en 2 pacientes (66,7 %) de DM, 1 caso de PM (33,3 %), en ambos casos se relacionó con artritis (100 %) (tabla 6).

Discusión
Las miopatías inflamatorias se caracterizan por presentar debilidad muscular proximal tanto escapular como pelviana, en ocasiones toma de la musculatura del cuello, además puede comprometer órganos del sistema que puede llegar a ser severa y provocar la muerte del paciente. A pesar de ser una enfermedad poco frecuente es importante su conocimiento debido a su complejidad y su característica de producción de auto anticuerpos específicos, marcadores muy útiles para el diagnóstico clínico, la clasificación y la predicción del pronóstico de estas, además de tener diferentes implicaciones en función de la edad, raza y factores ambientales.
Las variables sociodemográficas de los pacientes estudiados, demuestran que son similares a las series asiáticas,16 mesoamericanas,17 arábigas18 y población caucásica19 y algunos autores20 han encontrado un predominio del sexo femenino en proporción 4:1 y del color de piel no blanca.
En una investigación realizada por Smoyer-Tomic y otros en pacientes con MII, el sexo predominante fue el femenino con 2009 (71,3 %) pacientes, para una relación 2:1, el 42,9 % (n=220) eran afroamericanos, 2 087 (84,7 %) eran de procedencia urbana. El antecedente patológico personal más frecuente fue la hipertensión arterial en 1 153 enfermos (43,1 %) resultados que concuerdan con nuestro estudio, aunque la relación de la porción y color de la piel fue mayor que nuestra serie.21
Gonzalo Bello y otros en un estudio multicéntrico determinó anticuerpos en 77 pacientes con MMI, donde predominó la DM 71 %, de ellos la mayoría de sexo femenino (87 %), porcentaje algo superior a nuestra serie.22
La edad media de nuestros casos fue casi similar tanto para la DM como la PM (42,6 y 43,2, respectivamente), algo inferior a lo encontrado por Campo Voegeli,23 donde la media de edad de los 16 casos con MII estudiados fue de 53,6 años, con un predominio de los pacientes entre los 50 y 59 años. El sexo predominante fue el femenino con 10 pacientes, lo que representó 62,5 %, similar al nuestro. En el momento de ser incluidos en el estudio la enfermedad se consideró activa clínicamente en 12 enfermos (75 %) y en remisión en los otros 4 (25 %), que difiere con respecto al nuestro y podría deberse a que ellos lo hicieron de forma retrospectiva y nuestros pacientes lo determinamos en el estado actual que llevan tratamiento farmacológico que los ha mantenido en remisión. La HTA fue el antecedente patológico personal que más se presentó en el 25 % (n=4), resultados que se asemeja a los de esta serie.23
La mayoría de los pacientes evaluados por Kalluru y otros24 pertenecían al sexo femenino (12/20, 60 %), similar a nuestro estudio. Pero difiere en la edad media en el momento del diagnóstico que fue de 60 años, con un rango entre 54 y 87 año mayor al nuestro. Los anticuerpos fueron medidos en 17 enfermos, encontrándolos en 11 de ellos, lo que significó el 64,7 %, resultados superiores a nuestra investigación.24
De acuerdo a Maravi y otros25 en el estudio predominó el sexo femenino en el 80 % con una edad media de 53 años y tiempo de evolución de 8,9 años, en un 63 % se encontraron anticuerpos específicos y 33 % afectación pulmonar. Esto coincide con nuestro estudio en relación con el predominio en el sexo femenino, aunque en un porcentaje mayor al nuestro, pero difiere en la edad del comienzo de la enfermedad y la neumopatía intersticial fue superior a la nuestra. Con respecto al tiempo de evolución fue cercana a la nuestra, pero la diferencia puede estar relacionado con el tamaño de la muestra, ya que fue solo de 15 pacientes.
Nuestro estudio se relaciona con lo reportado por Marie26 un total de los 35 pacientes estudiados 13 (37,1 %) fueron hombres y 22 (62,9 %) mujeres, mostrando un predominio del sexo femenino en proporción 3:1. La asociación más frecuente, en cerca de un tercio de los pacientes, fue con enfermedades autoinmunes, en 17 % de los casos en relación con conectivopatías, lo cual difiere con el nuestro ya que solo estudiamos a las miopatías inflamatorias idiopáticas primarias no asociadas a enfermedades del tejido conectivo. En 20 % de los pacientes se identificaron otras enfermedades autoinmunes concomitantes, en la que destaca el hipotiroidismo, superior al nuestro dónde encontramos el 11,5 %.
Nuestro trabajo coincide con lo realizado por Laura Nuño y otros donde predomino el sexo femenino (74,1 %) y deferimos con ellos porque predominó en las caucásicos (83,5 %).26,27
En cuanto a las manifestaciones clínicas se destaca la elevada prevalencia de enfermedad pulmonar intersticial (29,9 %) encontrando daño pulmonar solo en la polimiositis y no en la dermatomiositis a diferencia a lo encontrado por Nuño y otros que fue superior a la nuestra.27
Dentro de las manifestaciones clínicas de compromiso sistémico presentadas por los pacientes, las más frecuentes fue la neumopatía intersticial en el 23,1 %, cifra inferior a lo encontrado en la investigación de Hochbert y otros donde los órganos afectados por la MII fueron el aparato respiratorio en 2 pacientes en forma de enfermedad pulmonar intersticial-, y el corazón - en 1 paciente, similar a nuestro estudio.28
Las manifestaciones orgánicas de la MII encontradas en el estudio de Xicará Sarpe que fueron la gastropatía erosiva en el 14,75 %, la esofagitis y artritis en el 4,91 % cada una de ellas, la enfermedad intersticial y la enfermedad cardiaca (trastornos del ritmo como una taquicardia supraventricular y un caso de disautonomía) en el 3,27 % cada una, resultados que no concuerdan con los de este trabajo.29
Laura Nuño y otros encontró que la enfermedad pulmonar intersticial en el 29,9 % superior a la nuestra. El 54,6 % de los pacientes presentaron algún factor de riesgo cardiovascular, principalmente dislipidemia, hipertensión arterial, diabetes mellitus.27
La enfermedad pulmonar en el 38 %, la enfermedad gastrointestinal en el 35 % y la enfermedad cardiaca en el 28 %, fueron las manifestaciones de compromiso general de la MII más frecuentes encontradas por Torres y otros en su serie, resultados que no concuerdan totalmente con los de este estudio pues nosotros no encontramos pacientes con afectación cardiaca y gástrica fue baja.30
En relación con la frecuencia de anticuerpos en nuestros estudios, encontramos mayor porciento de anticuerpos específico 48 % en relación al estudio realizado por Nuño y otros que se encontró un 30 % al igual a los anticuerpos asociados pero la muestra de ellos fu e superior a la nuestra.27
En cuanto a los anticuerpos específicos de miopatía, se encontraron en orden de frecuencia el Jo-1 en el 26,9 %, el Mi-2 en el 15,4 % y el SRP en el 5,8 % del total de las miopatías, en lo que respecta a los anticuerpos asociados el orden de frecuencia fue el Ro52 en el 21,3 %, el Ro 60 en el 11,5 %, el Ku en el 5,8 % y el PMScl en el 3,8 %, resultados algo similar al estudio de Nuño y otros.27
En nuestro estudio el anti Jo-1, es más frecuente a lo reportado por Lega JC y otros, que informan de una frecuencia del 6 % en las MMI. La frecuencia del anti-SRP en nuestra investigación se parece más a los de cohortes europeas que reportan una frecuencia de este anticuerpo entre el 6-8 %, y más común que en cohortes asiáticas que son de solo el 2 %.31,32
Los resultados de nuestro estudio son similares en cuanto al hallazgo de Ac específicos de miositis, aunque no concuerdan en el orden de frecuencia con lo encontrado en la serie de O’Callaghan y otros. En este último, los Ac específicos de miositis que en orden de frecuencia se encontraron fueron el Mi-2, seguido de Jo-1 y con respecto a los anticuerpos asociados, estos fueron positivos anti Ro en tres pacientes y anti SCL 70 en 2 pacientes con DM. Lo anterior difiere a nuestro estudio ya que se encontró el anti Ro en mayor número de pacientes y no anti SCL 70 debido a que nuestra investigación no incluyó miopatías asociadas a enfermedades del tejido conectivo porque este anticuerpo se encuentra en pacientes con esclerosis sistémica y el anticuerpo más frecuente fue el anti Jo1 seguido ala d Mi-2.33
Nuestro estudio difiere de Ernste34 debido a que en dicha investigación se buscó determinar la relación entre la presencia de anticuerpos específicos y asociados a miositis con las manifestaciones clínicas y se encontró una alta relación entre la presencia de estos con el desarrollo de manifestaciones clínicas, fundamentalmente el Jo1 y el Ro 52. Destacó entre ellas la neumopatía intersticial y se confirmó el Síndrome Antisintetasa, caracterizado por miositis, neumopatía intersticial, artritis, fenómeno de Raynaud, manos de mecánico. Esto concuerda con series de pacientes japoneses como la de Ikeda y otros y Hamaguchi y otros quienes hallaron también una importante relación entre los anticuerpos y el desarrollo de manifestaciones clínicas.27,35
El anti Mi-2 se asoció a las lesiones cutáneas características de las Dermatomiositis, así como los anticuerpos anti PM-ScL y anti Ku se asoció con cambios de la textura de la piel (endurecimiento) en dos pacientes tras varios años de evolución.
Predominaron los pacientes del sexo femenino, en la cuarta década de la vida, de color de piel no blanca y de procedencia urbana. Las manifestaciones clínicas más frecuentes fueron la neumopatía intersticial y la artritis. Los anticuerpos específicos de miopatías más frecuentes fueron el anti Jo-1, seguido por el anti Mi-2. Los anticuerpos asociados a miopatías más frecuentes fueron el Anti Ro 52 y anti Ro 60, predominaron ambos en la Dermatomiositis. El anticuerpo anti Jo-1 estuvo más relacionado con el daño pulmonar y el anti Mi-2 con las manifestaciones cutáneas, sin daño pulmonar.

