










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión impresa ISSN 0034-7531
Rev Cubana Pediatr vol.84 no.3 Ciudad de la Habana jul.-set. 2012
PRESENTACIÓN DE CASO
Hemosiderosis pulmonar idiopática
Idiopathic pulmonary hemosiderosis
MSc. Dra. Marlen Rivero González, Dra. Frances Seiglie Díaz, MSc. Dra. María del Carmen Luís Álvarez, MSc. Dra. Odette Pantoja Pereda, MSc. Dr. Alberto Arencibia Núñez
Hospital Pediátrico Universitario "William Soler". La Habana, Cuba.
RESUMEN
Se denomina hemosiderosis pulmonar a los procesos caracterizados por depósitos anormales de hemosiderina en el parénquima pulmonar, secundarios a sangrados alveolares difusos y repetidos. Es una enfermedad de causa desconocida, poco frecuente, y en muchas ocasiones grave. En la mayoría de los pacientes se presenta en la primera década de la vida, sin predilección en cuanto a sexo. Se presenta una paciente de 7 años de edad, femenina, de piel blanca, con antecedentes de 22 ingresos desde la etapa de lactante por episodios recurrentes de dificultad respiratoria, interpretados como bronconeumonías, asociados a anemia aguda. Para el diagnóstico se realizó lavado broncoalveolar, y se observaron los macrófagos cargados de hemosiderina. La evaluación clínica y de laboratorio permitió excluir causas secundarias. Se instauró tratamiento con prednisona, con lo cual se logró una mejoría de la enfermedad. Se discuten los elementos clínicos, diagnósticos y terapéuticos de esta entidad.
Palabras clave: hemosiderosis pulmonar idiopática, anemia ferropénica, hemorragia alveolar difusa.
SUMMARY
Idiopathic pulmonary hemosiderosis is those processes characterized by anomalous depots of hemosiderin in the pulmonary parenchyma, secondary to diffuse and repeated alveolar bleedings. It is an unknown disease, uncommon and mostly severe. It occurs in the first decade of life of most of the patients, regardless of sex. Here is a 7 years-old patient, female, Caucasian, with a history of 22 hospitalizations since she was a baby, due to recurrent episodes of respiratory distress diagnosed as bronchial pneumonias associated to acute anemia. For the diagnosis of this disease, bronchoalveolar lavage was performed and hemosiderin-loaded macrophages were observed. The clinical and lab evaluation excluded secondary causes. She was treated with prednisone and she improved her condition. The clinical, diagnosing and therapeutic elements of this disease were discussed.
Key words: idiopathic pulmonary hemosiderosis, iron-deficiency anemia, diffuse alveolar hemorrhage.
INTRODUCCIÓN
La hemosiderosis pulmonar idiopática (HPI) es una enfermedad de causa desconocida, poco frecuente, y en muchas ocasiones grave. En la mayoría de los pacientes se presenta en la primera década de la vida, sin predilección en cuanto a sexo. Esta condición ocurre en ausencia de otras enfermedades asociadas a hemorragia pulmonar.1
La enfermedad se caracteriza por sangrado intraalveolar difuso, por lo que habrá manifestaciones dependientes de la anemia, y otras dependientes del daño al parénquima pulmonar. La enfermedad se puede presentar en su forma clásica, caracterizada por la tríada de anemia, hemoptisis e infiltrados pulmonares. En estos casos la hemoptisis puede ser de diferente cuantía, en ocasiones de gran magnitud, poner en riesgo la vida del paciente, y en otras ocasiones sin evidencia clínica.2 La otra modalidad de presentación es en forma insidiosa por pérdida crónica de sangre, que se manifiesta como anemia ferripriva, y resulta refractaria a la terapia con hierro. La sangre deglutida puede simular una hemorragia digestiva.2
En la fase aguda se puede presentar fiebre, taquicardia, polipnea, tos, dolor abdominal, hepatoesplenomegalia transitoria y leucocitosis. En la radiografía de tórax se observan imágenes pulmonares condensantes, especialmente en los hilios y en ambas bases, que duran de 2 a 3 días, y pueden confundirse con una neumonía, lo que hace retrasar el diagnóstico. Posteriormente se observa un patrón reticular intersticial, que se normaliza entre los 10 y 12 días del inicio.3
Además se presentan síntomas respiratorias recurrentes o crónicos, tales como, tos, disnea, sibilancias y cianosis. Al existir episodios repetidos de sangrado, en ambas modalidades se llega a la fibrosis pulmonar progresiva.4
Debido a que la hemorragia alveolar difusa puede ser la forma de presentación, o una de las manifestaciones de múltiples entidades, el pediatra debe realizar un examen clínico minucioso para detectar síntomas y signos de enfermedad sistémica (sinusitis, vasculitis leucocitoclástica cutánea, iridociclitis, sinovitis, glomerulonefritis, etc.). Las entidades que deben descartarse ante un paciente con sospecha de hemosiderosis pulmonar son las siguientes (cuadro):5
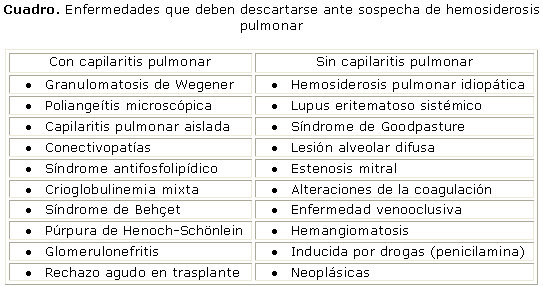
Aunque el diagnóstico de la HPI se realiza por exclusión de otras causas, lo más importante es la sospecha clínica ante un paciente con las manifestaciones clínicas descritas. Nuestro propósito es presentar la experiencia de un caso en nuestro servicio, y describir las dificultades en el diagnóstico, su evolución clínica y las modalidades terapéuticas utilizadas.
PRESENTACIÓN DEL CASO
Se presenta una paciente de 7 años de edad, del sexo femenino, con antecedentes de 22 ingresos desde la etapa de lactante por episodios recurrentes de dificultad respiratoria, interpretados como bronconeumonías y piodermitis a repetición. En varias ocasiones fue transfundida por presentar anemia severa asociada a los procesos respiratorios.
En julio de 2009 ingresa en nuestro servicio muy decaída, con fiebre, tos productiva, incremento de las secreciones respiratorias y esputos hemoptoicos. Al examen físico se detecta palidez cutáneo mucosa, íctero ligero, taquicardia (126 × min), soplo sistólico 2/6, polipnea (30 × min), tiraje intercostal y subcostal, ortopnea, abundantes ruidos transmitidos y estertores subcrepitantes en ambos campos pulmonares, y hepatoesplenomegalia. Además, presentaba uñas en vidrio de reloj, dedos en palillo de tambor, secreciones amarillas y costrosas en fosas nasales, así como lesiones cutáneas de piodermitis en resolución. El cuadro de la paciente progresó rápidamente al distrés respiratorio, con profundización de la anemia y deterioro clínico.
En los complementarios realizados se detecta anemia severa (62 g/L) y reticulocitosis de 10,4 %, leucocitos en 5,4 × 109/L con diferencial normal, y trombocitosis ligera (478 × 109/L). Las constantes corpusculares mostraban una anemia microcítica hipocrómica, con un volumen corpuscular medio (VCM) de 78 fentilitros (fL), que se confirmó en el examen de la lámina de periferia.
El estudio de hierro fue compatible con una anemia ferropénica (hierro sérico: 5,2 microsmol/litro; capacidad total de saturación de transferrina: 80 microsmol/litro; índice de saturación: 0,06). La prueba de Coombs directa negativa y la electroforesis de Hb AA con resistencia osmótica aumentada descartaron otras posibilidades diagnósticas.
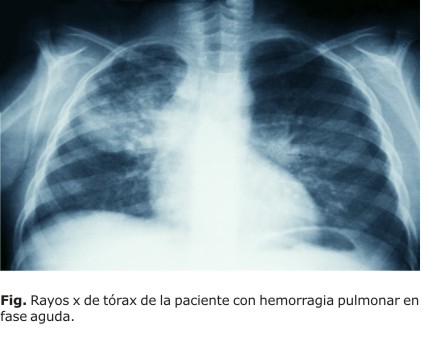
En la radiografía de tórax al ingreso se observaba hemorragia pulmonar (figura). En el ultrasonido de abdomen se demuestra hepatomegalia de 3 cm y esplenomegalia discreta. En la tomografía axial computarizada (TAC) de pulmón simple y con contraste, se describe la presencia de bronquiectasias en el pulmón derecho y cambios fibróticos en ambas bases pulmonares.
Ante estos elementos clínicos y de laboratorio se sospechó una hemosiderosis pulmonar, y se realizaron en 2 oportunidades lavado gástrico buscando siderófagos. Ante la negatividad de la prueba, se realizó lavado broncoalveolar (LBA), en el que se observaron los macrófagos cargados de hemosiderina en proporción mayor al 20 %.
Para descartar otras enfermedades sistémicas se realizaron exámenes para determinar anticuerpos antinucleares (ANA), anticitoplasma de neutrófilo (ANCA), antiDNA de doble cadena y antitransglutaminasa (ATG); inmunocomplejos circulantes (ICC), anticuerpos antiplaquetarios, anticoagulante lúpico, factor reumatoideo y dosificación de crioglobulinas, los cuales fueron negativos. En el perfil hepático tuvo elevación de transaminasa glutámico pirúvica (TGP, 119,7 UI), mientras la transaminasa glutámico oxalacética (TGO) fue normal (9,6 UI). El perfil renal, el proteinograma, el estudio del sedimento urinario y los exámenes de heces fecales fueron normales.
La serología para virus de hepatitis B y C fue negativa. En el estudio inmunológico se detectó una inmunodeficiencia celular con niveles bajos de linfocitos T CD4. La gasometría arterial fue normal, y la prueba de función respiratoria mostró un patrón restrictivo moderado.
Se inicia tratamiento con antimicrobianos, factor de transferencia, y ante la sospecha de una HPI, se impone tratamiento con prednisona 2,5 mg/kg/día y fumarato ferroso para contrarrestar la deficiencia de hierro. La paciente se recupera progresivamente, y es egresada con prednisona oral a 1 mg/kg/día. Además, se suprimió el consumo de lácteos como uno de los posibles desencadenantes.
Al alta tenía mejoría del estado general, no presentaba dificultad respiratoria, sin estertores a la auscultación pulmonar y desapareció la hepatoesplenomegalia. Tenía una Hb 112 g/L, reticulocitos normales, leucocitos en 9 × 109/L, con conteo diferencial normal, y eritrosedimentación en 8 mm/hora. En la consulta de seguimiento 3 meses después se mantenía con disnea ligera a los esfuerzos, pero sin anemia ni recurrencias del cuadro agudo.
DISCUSIÓN
La HPI es una entidad que se caracteriza clásicamente por la tríada de anemia, hemoptisis e infiltrados intersticiales pulmonares, por lo que es necesario para el diagnóstico la presencia de hemosiderófagos > 20 % en las secreciones bronquiales. Además, se debe demostrar la ausencia de otras causas que justifiquen el sangrado alveolar.6,7
La mayoría de las manifestaciones clínicas están relacionadas con la presencia de sangre en los alveolos, y con los efectos de la pérdida sanguínea crónica. Los síntomas son los de una enfermedad pulmonar recurrente o crónica, y consisten en tos, hemoptisis, disnea, sibilancias, y a veces, cianosis, acompañados de fatiga y palidez. La tos puede ser provocar esputo hemoptoico, o simplemente vomitar grandes cantidades de sangre.8
El caso clínico que se presenta tiene antecedentes de 22 ingresos por procesos respiratorios interpretados como bronconeumonías. Generalmente los episodios de sangrado alveolar se acompañan de fiebre, tos productiva, disnea, leucocitosis y alteraciones radiológicas; por tanto, es muy común que se interprete como un proceso infeccioso bacteriano.9,10
La anemia típica es microcítica e hipocrómica, y las concentraciones séricas de hierro son bajas, pues el sangrado crónico que se produce en la HPI deprime las reservas de hierro. Además, puede haber elevaciones de la bilirrubina y el urobilinógeno debido al metabolismo de la hemoglobina que se encuentra en el espacio extravascular.11 El patrón de anemia de la paciente fue compatible con una deficiencia de hierro, y la presencia de reticulocitosis evidenciaba la respuesta medular ante una pérdida aguda de sangre.
La HPI debe ser considerada en el diagnóstico diferencial de la anemia por deficiencia de hierro que no responde a la ferroterapia; además, la sospecha clínica aumenta notablemente cuando la ferropenia se asocia a reticulocitosis.12 El diagnóstico de certeza se completa al demostrar la presencia de hemosiderina en los macrófagos procedentes de las vías respiratorias. Estos se pueden obtener de extensiones de esputo, material obtenido por aspiración gástrica o traqueal, mediante LBA, o por biopsia pulmonar.1,13
La sensibilidad de las muestras es mayor en el LBA y la biopsia pulmonar. Esta última se puede realizar por toracotomía; sin embargo, se prefiere la video-toracoscopia, que es una técnica por mínimo acceso que disminuye las complicaciones peri y posoperatorias. La ventaja de la biopsia pulmonar es que se puede analizar el tejido pulmonar y realizar estudios de inmunofluorescencia para definir el diagnóstico etiológico de la hemorragia pulmonar.5,14
Las secreciones obtenidas del material gástrico tienen mayores índices de falso negativo, pero es un método menos invasivo que se utiliza en niños pequeños y pacientes con deterioro del estado clínico.1,13 En nuestra paciente la aspiración gástrica resultó negativa en 2 ocasiones; sin embargo, al mejorar su estado clínico, se realizó LBA que demostró la presencia de macrófagos cargados de hemosiderina y permitió descartar las causas infecciosas.
El deterioro de la función pulmonar es una de las complicaciones más temidas de esta enfermedad. El sangrado recurrente, con el cual se deposita hemosiderina en el tejido pulmonar, produce la destrucción de las fibras elásticas, que conduce a la fibrosis intersticial pulmonar. El tiempo de instalación de la fibrosis es variable, pero puede ser tan precoz como a los 2 meses de los primeros síntomas.15 En el caso que presentamos se encontraron elementos clínicos (uñas en vidrio de reloj y dedos hipocráticos), radiológicos y de función ventilatoria, que reflejaban la insuficiencia respiratoria crónica secundaria a la fibrosis pulmonar.
La baja frecuencia de la enfermedad y la expresión incompleta de las manifestaciones clínicas en algunos pacientes, provoca que el tiempo entre los primeros síntomas y el diagnóstico sea muy variable. Muchos casos han sido son catalogados como "asma", "neumonías a repetición" o "anemias refractarias", muchos años antes de establecer el diagnóstico de HPI. El tiempo de latencia antes del diagnóstico de nuestra paciente fue de 6 años. Una serie de casos en Chile, reportó una latencia de 5 años en el 60 % de los pacientes.3
La etiología de la HPI no está bien definida. Algunos autores señalan factores genéticos, mientras otros apoyan la influencia de elementos ambientales. Se ha reportado el aumento de la incidencia en individuos de la misma familia;8 sin embargo, la ausencia de la enfermedad en un gemelo idéntico de un paciente con HPI demuestra que el factor genético no es suficiente.13 Observaciones no validadas en estudios epidemiológicos han atribuido a los plaguicidas un papel etiológico dudoso.16 En nuestro caso no se documentaron antecedentes familiares previos, y aunque la paciente procedía del área rural, no refirieron contacto con tóxicos ambientales.
La etiología inmunológica de la enfermedad es la más probable. Se demostrado la asociación de la HPI con la alergia a la leche de vaca (síndrome de Heiner),17 así como con la enfermedad celiaca.18 Además, durante el seguimiento prolongado se presenta alguna enfermedad autoinmune en el 25 % de los pacientes.19 En nuestra paciente no se encontraron evidencias de autoinmunidad, pero presentó inmunodeficiencia celular, que puede provocar la disregulación del sistema inmune y desencadenar fenómenos autoinmunes.20
El tratamiento de elección son los esteroides. En la fase aguda se pueden administrar bolos con dosis altas de metilprednisolona, además de las medidas de soporte con oxígeno y ventilación asistida según grado de gravedad, así como broncodilatadores y antibióticos para evitar la coinfección.21
En la fase crónica de la enfermedad, la prednisona (1-2 mg/kg/día) es el fármaco de mejores resultados. La combinación con azatioprina (2 mg/kg/día) ofrece mejores resultados en los casos graves con recurrencias frecuentes. Es recomendable la inhalación de budesonide 200 microgramos 2 veces al día. Se han utilizado otros inmunosupresores como la ciclofosfamida, la 6-mercaptopurina, y la hidroxicloroquina en casos aislados. Es aconsejable suprimir la leche de vaca y cualquier otro antígeno demostrado en los estudios inmunológicos.1,7,13,15,22
Aproximadamente, la mitad de los niños fallecen en 1 a 5 años, habitualmente de hemorragia pulmonar aguda o insuficiencia respiratoria progresiva. La enfermedad tiene un curso más severo en los niños, sobre todo, si no se instaura el tratamiento específico de forma precoz.3,19
Se concluye que la HPI es una entidad poco frecuente que cursa con anemia, hemoptisis e infiltrados pulmonares. La sospecha clínica debe guiar al diagnóstico precoz, para instaurar el tratamiento específico que limite la progresión de la enfermedad y las secuelas pulmonares a largo plazo.
REFERENCIAS BIBLIOGRÁFICAS
1. Ioachimescu OC, Sieber S, Kotch A. Idiopathic pulmonary haemosiderosis revisited. Eur Respir J. 2004 Jul;24(1):162-70.
2. Ferrari GF, Fioretto JR, Alves AF, Brandao GS. Idiopathic pulmonary hemosiderosis: case report. J Pediatr (Rio J). 2000 Mar-Apr;76(2):149-52.
3. Rubilar L, Maggiolo J, Girardi G, González R. Hemosiderosis pulmonar idiopática. Neumol Pediatr. 2006;1(1):30-3.
4. Frankel SK, Cosgrove GP, Brown KK. Small vessel vasculitis of the lung. Chron Respir Dis. 2005;2(2):75-84.
5. Gomez-Roman JJ. Diffuse alveolar hemorrhage. Arch Bronconeumol. 2008 Aug;44(8):428-36.
6. Alsina L, García S. Neumonías recurrentes y persistentes. Manejo diagnóstico por el pediatra. Acta Pediatr Esp. 2005;63:96-100.
7. Kamienska E, Urasinski T, Gawlikowska-Sroka A, Glura B, Pogorzelski A. Idiopathic pulmonary hemosiderosis in a 9-year-old girl. Eur J Med Res. 2009 Dec 7;14 Suppl 4:112-5.
8. Gencer M, Ceylan E, Bitiren M, Koc A. Two sisters with idiopathic pulmonary hemosiderosis. Can Respir J. 2007 Nov-Dec;14(8):490-3.
9. Tardio-Torío E, Sánchez-Sánchez E, Cruz-Hernández M. Bronconeumopatías diversas. En: Cruz-Hernández M, editor. Tratado de pediatría. Madrid: Océano/Ergon; 2007. p. 1347-58.
10. Sheares BJ. Recurrent pneumonia in children. Pediatr Ann. 2002 Feb;31(2):109-14.
11. Andrews NC. Iron Deficiency and Related Disorders. En: Greer JP, editor. Wintrobe's Clinical Hematology. 12 ed. Philadelphia: Lippincott Williams & Wilkins; 2009. p. 811-30.
12. Valdés-Martín S, Gómez-Vasallo A. Temas de pediatría. La Habana: Editorial Ciencias Médicas; 2006. p. 350-62.
13. Zhang X, Wang L, Lu A, Zhang M. Clinical study of 28 cases of paediatric idiopathic pulmonary haemosiderosis. J Trop Pediatr. 2010 Dec;56(6):386-90.
14. Schwarz MI, Brown KK. Small vessel vasculitis of the lung. Thorax. 2000 Jun;55(6):502-10.
15. Yao TC, Hung IJ, Jaing TH, Yang CP. Pitfalls in the diagnosis of idiopathic pulmonary haemosiderosis. Arch Dis Child. 2002 Jun;86(6):436-8.
16. Cassimos CD, Chryssanthopoulos C, Panagiotidou C. Epidemiologic observations in idiopathic pulmonary hemosiderosis. J Pediatr. 1983 May;102(5):698-702.
17. Torres MJ, Giron MD, Corzo JL, Rodriguez F, Moreno F, Perez E, et al. Release of inflammatory mediators after cow's milk intake in a newborn with idiopathic pulmonary hemosiderosis. J Allergy Clin Immunol. 1996 Dec;98(6 Pt 1):1120-3.
18. Agarwal R, Aggarwal AN, Gupta D. Lane-Hamilton syndrome: simultaneous occurrence of coeliac disease and idiopathic pulmonary haemosiderosis. Intern Med J. 2007 Jan;37(1):65-7.
19. Le Clainche L, Le Bourgeois M, Fauroux B, Forenza N, Dommergues JP, Desbois JC, et al. Long-term outcome of idiopathic pulmonary hemosiderosis in children. Medicine (Baltimore). 2000 Sep;79(5):318-26.
20. Hoffman R, Benz EJ, Shattil SJ, Furie B, Silberstein LE, McGlave P, et al. Severe combined immunodeficiencies. En: Hoffman R, editor. Hematology: Basic Principles and Practice. 5th ed. Philadelphia: Churchill Livingstone; 2008. p. 517-49.
21. Vinodh BN, Sharma SK, Mukhopadhyay S, Ray R. Idiopathic pulmonary haemosiderosis: two case reports. Indian J Chest Dis Allied Sci. 2006 Jan-Mar;48(1):75-7.
22. Garcia-Luzardo Mdel R, Aguilar-Fernandez AJ, Cabrera-Roca G. Idiopathic pulmonary haemosiderosis in childhood: a good response to systemic steroids, inhaled hydroxychloroquine and budesonide. Arch Bronconeumol. 2010 Nov;46(11):612-3.
Recibido: 21 de noviembre de 2011.
Aprobado: 7 de marzo de 2012.
Marlen Rivero González. Hospital Pediátrico Universitario "William Soler". San Francisco esquina Perla, reparto Altahabana, municipio Boyeros. La Habana, Cuba. Correo electrónico: marlenrg@infomed.sld.cu
