










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La distrofia miotónica tipo 1(DM1), enfermedad de Steinert o distrofia miotónica de Steinert (DMS) es una enfermedad autosómica dominante producida por la expansión de tripletes CTG en la región no codificante del gen DMPK (proteincinasa de la distrofia miotónica), localizada en el brazo largo del cromosoma 19 (19q13.3). Por tanto, el riesgo que tiene cada hijo de un paciente de heredar la mutación es de 50 %. La penetrancia (fenómeno biológico, cuyo término se emplea para referirse a la expresión fenotípica de un gen mutado) es muy alta, cercana a 100 % a los 50 años de edad, cuando se consideran todas las manifestaciones clínicas de la enfermedad.1
La gravedad del fenotipo del hijo dependerá del tamaño del fragmento CTG que herede. Al ser inestable la transmisión de este fragmento, con tendencia a incrementar el número de tripletes CTG que pasan a la descendencia, los hijos que hereden la mutación suelen presentar formas más graves que sus padres, fenómeno conocido como anticipación clínica.2
La distrofia miotónica congénita (DMC) es la forma más grave de DMS y tiene una incidencia estimada a nivel mundial de 3-15:100 000 nacidos vivos. La transmisión es, en más de 90 % de los casos, de origen materno por probable inestabilidad meiótica aumentada durante la ovogénesis, en relación con la espermatogénesis.3
La forma congénita, se reconoce por presentar las características de una secuencia de acinesia-hipocinesia fetal (SAF). Predominan los signos dismórficos faciales y de extremidades, a ello se suma la hipotonía neonatal e insuficiencia respiratoria aguda, con baja expectativa de supervivencia. Se presenta retraso en el desarrollo psicomotor tanto de la habilidad motora gruesa como en la fina. La discapacidad intelectual, severa o profunda, está siempre presente en los casos de DMS que se inician clínicamente al nacer. Durante el embarazo es característico: la disminución de los movimientos fetales, polihidramnios y presentación en pelviano, por lo que la mayoría de los partos se llevan a cabo por cesárea. La tasa de mortalidad fetal y perinatal asciende a 28 % y la esperanza de vida en esta forma no supera los 30 años de edad. La principal causa de muerte es la insuficiencia respiratoria grave.3,4
A continuación, se describen cuatro casos, con expresión clínica de distrofia miotónica congénita, que constituye el objetivo de este trabajo.
Presentación de casos
Los 4 pacientes, que conforman la serie presentada, tenían depresión neonatal severa, hipotonía, todos tenían antecedentes familiares por línea materna. La prevalencia fue de 12,6 × 100 000 nacidos vivos (tabla).
Tabla Descripción de los casos con expresión clínica de distrofia miotónica congénita
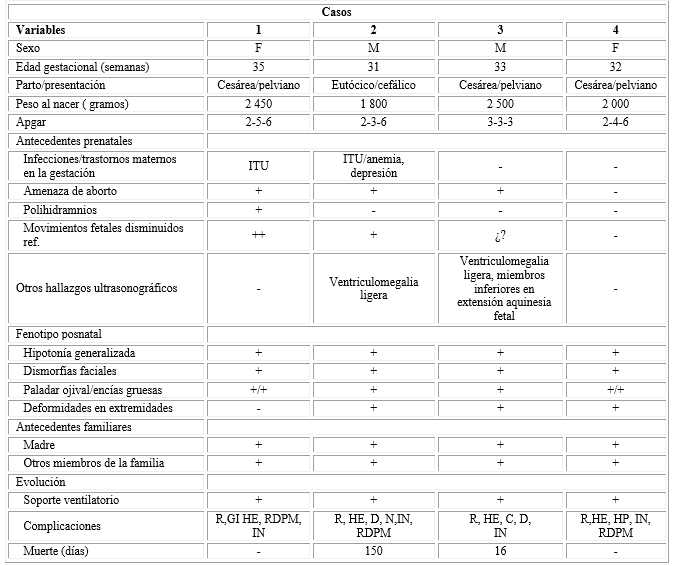
Prevalencia: 12,6 × 100 000 nacidos vivoM: masculino; F: femenino; ITU: infección del tracto urinario; R: respiratorio; GI: gastrointestinal; HE: trastornos hidroelectrolíticos; RDPM: retardo del desarrollo psicomotor; IN: infecciones; D: diafragmático; N: neumomediastino; C: cardiovascular; HP: hemorragia pulmonar.
Discusión
La enfermedad de Steinert congénita es la forma más grave de distrofia miotónica, caracterizada fundamentalmente por hipotonía muscular y dificultades para la alimentación.5En los casos con distrofia miotónica congénita es muy frecuente el parto distócico. Lo anterior es debido a la asociación de presentación en pelviano, transverso (por la hipomovilidad fetal) a lo cual se suma que las madres de estos niños también padecen la enfermedad, en estas hay mayor afectación de la musculatura uterina que produce contracciones de menor intensidad de lo habitual y que pudiera conducir a hemorragias después del parto. Todas estas condicionantes también constituyen causas de los partos distócicos (cesárea).5,6,7) Lo anterior coincide con los casos presentados en este trabajo puesto que describe la mayor frecuencia de cesárea.
Es habitual encontrar bajas puntuaciones de Apgar en estos recién nacidos, por debajo de 9 puntos al nacer y a los 5 minutos, lo cual coincide en todos los casos de la presente serie de casos, que tienen menor vitalidad dada la afectación muscular de base, de manera general en ellos se presenta menor puntuación del tono, reflejos, así como de los movimientos respiratorios y por tanto, del color. La asfixia perinatal puede ser la causa que origina la ventriculomegalia como hallazgo más habitual. En la mitad de los casos de la presente investigación se registra este signo, que también fue observado desde la etapa prenatal.6,7
En esta serie de casos, la hipomovilidad fetal fue referida en el 75 % de los casos, de estos solo en el 50 % hubo polihidramnios y en el 100 % se desencadenó un parto pretérmino. El polihidramnios parece ser una de las causas responsables de la relativa frecuencia de partos prematuros en la DMC. 6,7
Los aspectos dismorfológicos de la DMC se basan en un mecanismo fisiopatológico de inmovilidad/hipomovilidad fetal condicionado por la enfermedad muscular, cuyo cuadro clínico completo es conocido como secuencia de aquinesia/hipoquinesia fetal (SAF).6,7,8,9
La falta de movimientos origina la presencia de contracturas articulares múltiples (artrogriposis) y deformidades esqueléticas principalmente pie equino varo, tal y como se presentó en 75 % de los casos de esta serie. Asimismo, y secundario a la falta de la succión, deglución y masticación, se presentan las dismorfias craneofaciales como la retrognatia, facies sugestiva poco expresiva y alargada, paladar ojival, labio superior en forma de V invertida. Todos los elementos anteriores estaban presentes en todos los casos de este estudio.6,10
La enfermedad ha llegado a ser considerada una de las enfermedades con mayor variedad fenotípica que existe.1,2
Se hereda de forma autosómico dominante y además se produce el fenómeno de anticipación, y la penetrancia es mayor en generaciones sucesivas y, por tanto, formas más graves.11,12) En el presente estudio la entidad se comportó igual a lo registrado para esta forma de herencia.
La enfermedad, tiene una expresividad variable debida a la expansión inestable del triplete CTG en gen DMPK. Los afectados de la forma congénita tienen hasta 2500 repeticiones. Se ha relacionado el número de repeticiones del triplete con el grado de afectación de la enfermedad, ya que a más repeticiones la afectación suele ser mayor y más temprana; también se ha descrito la relación de mayor severidad fenotípica cuando la transmisión es por vía materna.11,12,13) En el 100 % de los casos que se presentan la transmisión es materna.
El diagnóstico de la entidad, es sospechado por la clínica y presencia de la enfermedad en la madre. La confirmación definitiva es por estudio de genética molecular. La electromiografía, no suele ser útil en el período neonatal.5) En la serie de casos que se presenta no fue posible realizar el estudio molecular dado la no disponibilidad de este tipo de estudio en Cuba.
Las complicaciones más notificadas son las alteraciones respiratorias. La hipotonía generalizada, las alteraciones faciales provocan problemas de insuficiencia respiratoria. La afección muscular de la DMC, provoca una serie de alteraciones en el mecanismo de la respiración (ausencia de movimientos torácicos por afectación de los músculos intercostales y diafragmáticos, inmadurez pulmonar en los prematuros, hipoplasia pulmonar). La hipoplasia pulmonar es un factor pronóstico fundamental, que está presente en aquellos casos que hayan desarrollado en toda su expresión el espectro de la SAF.6,7,9 Las principales complicaciones que condujeron a morbilidad y mortalidad en el 100 % de los casos del presente estudio fueron las relacionadas con el sistema respiratorio, trastornos hidroelectrolíticos y las infecciones asociadas.
En el último informe periódico de Orphanet , se publica una prevalencia de la distrofia miotónica de Steinert de 6,7 a 12,5 × 100 000,14 Los datos publicados en este documento son estimaciones mundiales, o estimaciones europeas, son datos recopilados en bruto o extrapolaciones de estos a nivel mundial o europeo cuando no se sospecha un efecto fundador como causa de una enfermedad.14 En el mundo, se reporta una incidencia de 3-15:100 000 nacidos vivos.5 En Pinar del Río se registra en la DMC una prevalencia de 12,6 × 100 000 nacidos vivos.
La gravedad de la forma congénita de distrofia miotónica de Steinert requiere un seguimiento estrecho por un equipo multidisciplinar para detectar las posibles complicaciones descritas que empeoran la calidad de vida de los pacientes y sus familias, pudiendo llegar incluso a la muerte, especialmente durante el primer año de vida. Actualmente el asesoramiento genético es esencial para la planificación familiar y poder evitar la transmisión de la enfermedad a generaciones sucesivas.
Podemos concluir que en el período neonatal son importantes los antecedentes prenatales-perinatales de los pacientes con distrofia miotónica. Estos antecedentes, constituyen acontecimientos que forman parte de la secuencia de hipoquinesia fetal dada por la afectación neuromuscular intraútero. Los antecedentes familiares y sobre todo cuando la madre está afectada conducen a expresiones severas en la descendencia.

