










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
El cáncer de ovario de origen epitelial(CEO) ocupa el 6to lugar en incidencia y mortalidad a nivel mundial y es el más letal de todos los cánceres ginecológicos.En Cuba ocupa el quintolugar en incidencia dentro de la población femenina de 18 a 45 años de edad y la séptima causa de cáncer después de los 45 años. Se diagnostica de forma tardía, en las etapas III/IV, en más del 75 % de los casos y cursa de forma indolente más allá de la mitad de su historia natural.1-4
Los estándares terapéuticos en este tipo de cáncer no generan una supervivencia mayor del 50 % a los cinco años. Sin embargo, la inmunoterapia ha emergido como un pilar más de tratamiento,pero se requiere una mejor caracterización del infiltrado inflamatorio acompañante del tumor para lograr un impacto positivo mayor.5-6
El cáncer de ovario es inmunogénico y se relaciona convarias estirpes celulares que lo promueven,entre las que se encuentran: fibroblastos asociados al tumor, células madre mesenquimales y adipocitos asociados al ovario; así como células del sistema inmune: macrófagos asociados a tumor (M2), células mieloides derivadas supresoras y linfocitos T reguladores. Por otra parte, hay células linfocitarias que destruyen este tumorcomo los T citotóxicos, las asesinas naturales(NK, siglas en inglés) y asesinas naturales T(NKT, siglas en inglés). Además, existen otras como las células dendríticas cuyaacción dependerá de su polarización funcional.5-8
La citología es el método diagnóstico de elección para caracterizar los infiltrados celulares en los líquidos corporales y la histopatología en los tejidos sólidos. Estos métodos tienen limitaciones, como que solo examinan porciones de la muestra recolectada. El reporte de los estudios patológicos, excepto la inmunohistoquímica, solo reportan linfocitos y no se puede determinar su linaje ni fenotipo funcional.9
Lacitometría de flujo (CF) posee múltiples ventajas sobre los estudios citológicos e inmunohistoquímicos. Esta tecnología posee una alta sensibilidad y especificidad, muy superior a la citología; es un método objetivo de alta velocidad que puede analizar más de 10000 eventos/s. Además, permiteel análisis multiparamétrico sobre una sola célula, a diferencia de la inmunohistoquímica, que habitualmente solo mide entre unoatresantígenos por corte de tejido.10-13
Los inmunofenotipos linfocitarios actuales son el resultado de evaluaciones complejas que miden simultáneamente entre 6 y 17 antígenos, e incluso más. De esta forma se ha abundado en la complejidad del sistema inmune y el descubrimiento de cientos de subpoblaciones estructurales y funcionales distintas, precisamente debido al desarrollo de la CF. Por tal motivo, resulta desventajoso el empleo de técnicas que requieren muchos cortes de tejido de una biopsia o que, solo estudien de manera muestral un área muy reducida de un fluido, como lo hace la citología.10-13
El objetivo este trabajo fue diseñar y optimizar un panel multicolor para evaluarlos inmunofenotipos linfocitarios en sangre, ascitis y tejido tumoral ovárico, en pacientes con cáncer de ovario. Posteriormente, con la ampliación de estos resultados y la metodología propuesta, se podrá inmunofenotipar un mayor número de fenotipos que poseen un significado relevante en la biología del cáncer de ovario e incluso se podrá emplear para estudios linfocitarios en otras enfermedades.
Métodos
Se llevó acabo un diseño experimental, para la creación de un panel multicolor de CF y su evaluación en el laboratorio de Inmunología del Instituto Nacional de Oncología y Radiobiología de Cuba (INOR). Las principales etapas de trabajo fueron: selección de losantígenos y de los anticuerpos monoclonales conjugados con fluorocromos; diseño de procedimientos técnicos de tinción, creación, evaluación y optimización del panel de citometría y, análisis de los datos adquiridos. Primero se obtuvo el panel para sangre periférica y, a partir de este se hicieron los ajustes para muestras especiales: ascitis y tejido epitelial ovárico. Se siguieron algunas recomendaciones generales emanadas de la Guía para el uso de la CF en estudios inmunológicos.11
Obtención de las muestras
Se obtuvieron2mL de sangre periférica de 3 adultos, supuestamente sanos, para la titulación de los conjugados, ajuste de voltajes de fotodetectores, generar la matriz de compensación inicial y realizar los controles al panel. Posteriormente, se optimizó la metodología con 33 muestras de sangre de voluntarios sanos y, por último, se analizaron 2 mL de sangre periférica,2mL de líquido ascítico y un fragmento de tejido ovárico,de trespacientes operadas por el servicio de Ginecología del INOR, con el diagnóstico histopatológico de CEO.El líquido ascítico se colectódurante el acto quirúrgico, en tubo Vaccutainer con ácido etiléndiaminotetracético (EDTA) y el tejido ovárico fresco, en tubo cónico estérilde 15mL con solución salina al 0.9%.
Consideraciones éticas
Los sujetos y paciente accedieron voluntariamente a participar y firmaron el consentimiento informado. Se cumplieron los requisitos emanados de la Declaración de Helsinki.14 Se garantizó la confidencialidad de los datos obtenidos y su uso con fines exclusivos de la investigación.El estudio no implicó riesgos éticos para los pacientes ya que no fueron sometidos a experimentación y estuvo aprobado por el comité de ética del INOR.
Selección de los antígenos y conjugados
Se seleccionaron 11 antígenos en total, el CD45 que es considerado panleucocitario y otros típicos de linaje de subpoblaciones linfocitariasB, T y NK; así como el CD25 y HLA-DR,antígenosde activación celular.Para la elección de los fluorocromosse consideraron los factores que influyen en la calidad de los resultados. Por ejemplo, basados en la configuración del citómetro se utilizaron fluorocromos que tuvieran menor superposición espectral, entre canales vecinos o se encontraran alejados en su espectro de emisión. También se determinó que losmás brillantes se emplearan para antígenos de poca expresión o que por su patrón de expresión fuesen de tipo secundario o terciario; mientras que los menos luminosos se utilizaron para la detección de los antígenos primarios o de alta expresión en lamembrana celular.
Todos los conjugados empleados fueron Beckman Coulter, Francia. Quedaron definidos cuatro tubos policromáticos para el panel:
anti-CD45 KRO (Clon J33), anti-CD19 PC7 (Clon 89B.B4), anti-CD3 FITC (Clon UCHT1), anti-CD4 PB (Clon 13B8.2), anti-CD8 AA700 (Clon B9.11), anti-CD56 PE (Clon N901) (NKH-1) y anti-CD34 APC (Clon 581).
anti-CD45 KRO (Clon J33), anti-CD19 PC7 (Clon 89B.B4), anti-CD20 FITC (Clon B9E9), CD25 PC5.5 (Clon B1.49.9) y anti-HLA-DR PE (Clon Immu-357).
anti-CD45 KRO (Clon J33), anti-CD3 PC5 (Clon UCHT1), anti-CD4 PB (Clon 13B8.2), anti-CD8 AA700 (Clon B9.11), anti-CD56 PE (Clon N901) (NKH-1) y anti-CD16 FITC (Clon 3G8).
anti-CD45 KRO (Clon J33), anti-CD3 FITC (Clon UCHT1), anti-CD4 PB (Clon 13B8.2), anti-CD8 AA700 (Clon B9.11), anti-HLA-DR PE (Clon Immu-357) (NKH-1) y CD25 PC5.5 (Clon B1.49.9).
Titulación de los conjugados
Se emplearon las muestras de sangre periférica y se siguieron las recomendaciones del fabricante en los procedimientos de tinción (Beckman Coulter, Francia).De la concentración propuesta por el fabricante, se realizaron 4 diluciones por mezcla de volúmenes decrecientes de cada conjugado en volúmenes fijos de muestra.
Se utilizó elmétodo de lisis de hematíes sin lavado: a cada volumen del conjugadose leañadieron 100µL de sangre,se mezcló por 4 s, se incubó en cámara oscura por 20 min a temperatura del laboratorio; se añadió 1mL de buffer de lisis VersaLyseTM (BeckmanCoulter, Francia) e incubó por 30 min en las mismas condiciones del paso anterior. Por último, se procedió a la adquisición de la muestra por el citómetro.
Para cada concentración del conjugado se determinó el índice de tinción mediante la división de la mediana de intensidad fluorescencia (MIF) de la población positiva entre la MIF de la población negativa. Se seleccionaron los índices mayores a la menor concentración del conjugado ya que discriminan mejor entre poblaciones positivas y negativas. Además, se tuvieron en cuenta que los porcientos de las poblaciones seleccionadas se encontrasen dentro de los valores normales reportados en la literatura. Finalmente se creó un coctel con las titulaciones individuales para 15 determinaciones con validez para 30 días.
Construcción del panel
Con las concentraciones óptimas elegidas para cada fluorocromo se comenzó a construir el panel. Primero se utilizó una muestra con células sin teñir para establecer los voltajes y ganancias de los fotodetectores de dispersión de la luz anterógrada (FS, siglas en inglés) y lateral(SS, siglas en inglés) y así obtener un gráfico biparámetrico, típico de las tres subpoblaciones leucocitarias principales: linfocitos, monocitos y granulocitos. Se realizó la misma operación con los fotomultiplicadores para visualizar la señal negativa yajustar sus voltajes.
Para la generación de la matriz de compensación se utilizó el kit de microesferas de captura VersaCompTM (BeckmanCoulter, Francia). Se siguieron las especificaciones del fabricante para el montaje de las muestras y para su ejecución se empleó el planificador automático de protocolos del software Gallios (AutoSetup),
Controles de fluorescencia menos uno (FMO, siglas en inglés)
Se realizó FMO para eliminar la influencia de diferentes conjugados en un canal determinado y de esta forma poder establecer adecuadamente los niveles de corte para el marcador que corre por ese canal. Este procedimiento solo se realizó para los conjugados específicos de antígenos secundarios y terciarios. El montaje se hizo en leucocitos de sangre periférica y por el mismo procedimiento de lisis sin lavado. No obstante, en el diseño del protocolo de adquisición se crearon gráficos de control interno para verificar los niveles de corte.
Diseño de protocolos de adquisición y análisis de datos por citometría
Se empleó un citómetro Beckman Coulter Gallios de 10 colores con tres láseres: azul 488nm, rojo 638nm y violeta 405nm, con software Gallios v1.2 para la adquisición de los datos y Kaluza Analysis v1.5a para el análisis y reporte de resultados. El control de calidad diario se realizó con Flow-CheckTM Pro Fluorospheres, (Beckman Coulter, Francia) para comprobar la estabilidad de los sistemas ópticos y de fluidos. Se utilizaron Flow-SetTMPro Fluorospheres (Beckman Coulter, Francia), para la calibración de la dispersión de la luz y la intensidad de fluorescencia.
Adquisición de datos
Se diseñaron protocolos de adquisición por cada tubo del panel. Se creó una estrategia de ventanas manual y secuencial. En los casos de la sangre periférica se adquirió un mínimo de 50000 eventos totales. Mientras que para los otros tipos de muestras se determinó un mínimo de 5000 eventos en la ventana de selección de linfocitos. Se fijó una velocidad baja de adquisición de 10 µL/min para reducir las imprecisiones en el punto de interrogación. Los niveles de corte de expresión de los diferentes antígenos, se realizó por controles negativos y FMO como se describió anteriormente.Además, se utilizó la estrategia de confirmar los cortes por controles negativos internos,consistentes en la clonación de gráficos procedentes de poblaciones francamente negativas para el marcador evaluado en la misma muestra.
En la estrategia de ventanas se tuvieron en cuenta diferentes controles de calidad interna de los datos, que permiten monitorear la validez de los resultados en cada muestra. Por ejemplo,control de la estabilidad óptica y de fluidos con un gráfico de fluorescencia vs tiempo de un canal representativo para cada láser (CD3 FITC/FL1/Láser azul, CD8 AA700/FL7/Láser Rojo, CD45 KRO/FL10/Láser Violeta) y gráficos para eliminación de detritus o células apoptóticas y deeventos coincidentes.11
Además, se crearon gráficos de control de eventos positivos para monitorear el patrón de expresión de cada marcador según el tipo de la población leucocitaria, sin la posible interferencia de la estrategia de ventanas, pero conservando la lógica en reversa de los resultados sobre la población de linfocitos.
Una vez optimizado el panel parasangre periférica, se procedió a ajustarlo con las muestras de líquido ascítico y de tejido epitelial ovárico.Se realizaron las precisiones requeridas a los protocolos en lo referente a voltajes y matriz de compensación. La estrategia de ventana fue la misma. En el procesamiento de la muestra se realizaron modificaciones por las características de estas.
Líquido ascítico
Se realizó un lavado de los 2mL del líquido ascítico con 2mL de solución de lavado (SL) compuesta por: solución salina al 0.9% (SL) con suero fetal bovino inactivado (BSA, siglas en inglés) al 1% y EDTA al 1mM. Después se centrifugó a 400g y se eliminó el sobrenadante por decantación. Se resuspendió en 1mL de SL, con agitación por 4 s. La tinción fue realizada con los conjugados previamente retitulados en una suspensión de 50µL de células, en tubos de citometría de 12x75mm (Beckman Coulter, Francia). Se incubó en cámara oscura a temperatura de laboratorio por 20 min. Después se adicionaron 2mL de SL y se aplicó agitación por 4 s, seguido de centrifugación a 400g. Posteriormente se eliminó el sobrenadante por decantación y fue completado otro paso de lavado igual al anterior. Finalmente se reconstituyeron las células de cada tubo en 150µL de SL. Previo tamizado de la muestra con filtros de 100 µm, se analizó inmediatamente en el citómetro.
Tejido tumoral ovárico
Inicialmente se realizó la disgregación mecánica con bisturí No. 22 en placas de cultivo estériles. Una vez disgregado se le adicionaron 10mL de SL empleada para la ascitis. Esta suspensión fue transferida a través de un filtro de 100 µm a un tubo cónico de 15 mL. Se centrifugó a 400g y se eliminó el sobrenadante por decantación. Después se resuspendió en 1mL de SL. Se aplicó agitación por tres pasos de 4 s cada uno. Se dispensaron los conjugados en los tubos de citometría (Beckkman Coulter, Francia) con los volúmenes previamente retitulados de los conjugados. Después seadicionaron 100µL de las células resuspendidas a cada uno. La incubación fue en cámara oscura a temperatura de laboratorio por 20 min. Después se realizaron dos pasos de lavado con SL como se describió para el líquido ascítico. Finalmente, se resuspendieron las células de cada tubo de citometría en 150µL de SL. Se filtró nuevamente con filtros de 70µm y se analizó inmediatamente en el citómetro.
Resultados
Selección de los antígenos y conjugados
Los marcadores seleccionados permitieron inmunofenotipar linfocitos B, T y NKen los tres compartimentos. Así como algunas de sus subpoblaciones importantes en la biología del cáncer de ovario, como linfocitos T cooperadores, citotóxicos, dobles negativos y dobles positivos para CD4 y CD8;NKT, NK secretoras y citotóxicas, linfocitos T activados y T reguladoras. El CD25 y HLA-DR se seleccionaron por ser los antígenos de la fase celular de activación tardíade los linfocitos T.
Titulación
Las concentraciones óptimas encontradas,estuvieron por debajo de las recomendadas por el fabricante y existieron variaciones de un conjugado a otro (Tabla 1). Alrededor del 60% de los conjugados mostraron el mayor índice de tinción con la cuarta parte de la concentración establecida por el fabricante.
Tabla 1 Conformación final del panel según los antígenos y fluorocromosseleccionados con su volumen óptimo de titulación
| Láser | Azul 488nm | Rojo 638nm | Violeta 405nm | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Panel | Fluorósforo | FITC | PE | PC5/PC5.5 | PC7 | APC | AA700 | Pacific Blue | KRO |
| Tubo 1 | Anticuerpo | CD3 | CD56 | - | CD19 | CD34 | CD8 | CD4 | CD45 |
| Volumen (µL) | 7,5 | 5 | - | 2,5 | 2,5 | 2,5 | 2,5 | 2,5 | |
| Tubo 2 | Anticuerpo | CD20 | HLA-DR | CD25 | CD19 | - | - | - | CD45 |
| Volumen (µL) | 7,5 | 5 | 2,5 | 2,5 | - | - | - | 2,5 | |
| Tubo 3 | Anticuerpo | CD16 | CD56 | CD3 | - | - | CD8 | CD4 | CD45 |
| Volumen(µL) | 7,5 | 5 | 2,5 | - | 2,5 | 2,5 | 2,5 | ||
| Tubo 4 | Anticuerpo | CD3 | HLA-DR | CD25 | - | - | CD8 | CD4 | CD45 |
| Volumen (µL) | 7,5 | 5 | 2,5 | - | - | 2,5 | 2,5 | 2,5 | |
Leyenda: Fluorocromos.FITC: isotiocianato de fluoresceína, PE: Ficoeritrina, PC5: ficoeritrina-cianina 5, PC5.5: ficoeritrina-cianina 5.5, PC7: ficoeritrina-cianidina 7, APC: aloficocianina, AA700: aloficocianina-Alexa Fluor-700, KRO: Krome Orange.
CD: Clúster de diferenciación
Un ejemplo del procedimiento realizado para seleccionar la concentración óptima se observa en la Fig. 1.
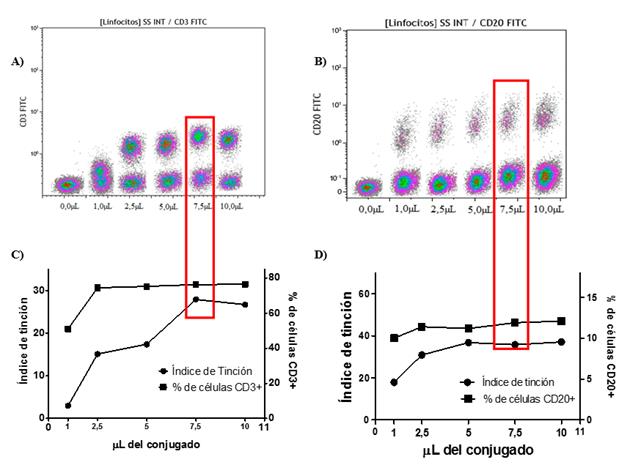 A y B: concatenación de imágenes de densidades para losantígenos CD3 y CD20, respectivamente. Datos analizados en Kaluza. C y D: distribución del índice de tinción según los volúmenes dispensados de los conjugados anti CD3 y anti CD20, respectivamente. El índice se obtuvo por el cociente entre la mediana de intensidad de fluorescencia (MIF) de la población positiva entre la MIF de la población negativa.Además, se muestran los porcentajes de laspoblaciones de linfocitos T y B, según los volúmenes de conjugados.Los recuadros rojos muestran los volúmenes que se adoptaron para la construcción final del panel. Datos analizados en GraphPadPrism 7.
A y B: concatenación de imágenes de densidades para losantígenos CD3 y CD20, respectivamente. Datos analizados en Kaluza. C y D: distribución del índice de tinción según los volúmenes dispensados de los conjugados anti CD3 y anti CD20, respectivamente. El índice se obtuvo por el cociente entre la mediana de intensidad de fluorescencia (MIF) de la población positiva entre la MIF de la población negativa.Además, se muestran los porcentajes de laspoblaciones de linfocitos T y B, según los volúmenes de conjugados.Los recuadros rojos muestran los volúmenes que se adoptaron para la construcción final del panel. Datos analizados en GraphPadPrism 7.Fig.1. Curvas de titulación de los conjugados.
En el CD45, que no posee población negativa en sangre periférica, se adoptó la concentración que mejor discriminó entre granulocitos, monocitos y linfocitos. Mientras que, en el líquido ascítico y las muestras de tejido ovárico, se calculó el índice de tinción para CD45 a partir de las células epiteliales como señal negativa.
No existieron afectaciones de los porcentajes de subpoblaciones linfocitarias con las concentraciones optimizadas de los conjugados.
Evaluación del panel diseñado
Construcción del panel: primero se adicionaron conjugados simples y posteriormente se adicionaron los conjugados con fluoróforos de tipo tándem. Se creó un coctel por cada tubo para una duración de 30 días y volumen proporcional para 15 determinaciones.
FMO: se empleó la estrategia de identificarel patrón de fluorescencia basal de los leucocitos normales sin teñir, para establecer los niveles de corte para cada marcador. Posteriormente se realizó la FMO para el CD56 PE (en poblaciones NK, NKT) y el HLA-DR PE y CD25 PC5 para población T. Adicionalmente, se diseñaron controles negativos internos para los tres marcadores antes expuestos y reforzar la precisión del nivel de corte. Se construyeron a partir de granulocitos que no expresan dichos antígenos.Solo se encontraron discretas interferencias en cuanto al nivel de corte de las poblaciones NKT y T activadas al analizar el tejido ovárico y se realizaron los ajustes establecidos por FMO y control interno. (Fig. 2)
Los controles isotípicos no fueron requeridos, pues no existieron marcajes intracelulares y se establecieron controles negativos para cada muestra. No se encontraron diferencias de ruido de fondo entre los protocolos que se realizaron sin lavados y en los que se emplearon pasos de lavado.
 A: Fluorescencia global vs dispersión lateral de la luz (SS, por sus siglas en inglés) por cada antígeno. Se utilizó para todos los tubos. B: Lógica secuencial de ventanas para eliminar loseventos coincidentes, detritus celulares y hematíes resistente a la lisis, selección de loslinfocitos por CD45+ vs SS, así como la identificación de lassubpoblaciones de interés. C: Selección de lassubpoblaciones sobre linfocitos B. D: Subpoblaciones de linfocitos T activados.Los gráficos encerrados en líneas de puntos con flechas, fueron seleccionados de la población de granulocitos para establecer los niveles de corte para los antígenos secundarios y terciarios, como controles negativos internos. Diseñado en software Kaluza Análisis V1.5a.
A: Fluorescencia global vs dispersión lateral de la luz (SS, por sus siglas en inglés) por cada antígeno. Se utilizó para todos los tubos. B: Lógica secuencial de ventanas para eliminar loseventos coincidentes, detritus celulares y hematíes resistente a la lisis, selección de loslinfocitos por CD45+ vs SS, así como la identificación de lassubpoblaciones de interés. C: Selección de lassubpoblaciones sobre linfocitos B. D: Subpoblaciones de linfocitos T activados.Los gráficos encerrados en líneas de puntos con flechas, fueron seleccionados de la población de granulocitos para establecer los niveles de corte para los antígenos secundarios y terciarios, como controles negativos internos. Diseñado en software Kaluza Análisis V1.5a.Fig. 2. Estrategia de ventanas sobre muestra de sangre periférica.
Diseño de protocolos de adquisición y análisis de datos por citometría
Lo voltajes y ganancias de los fotodetectores se realizaron con células no marcadas y se calibró con las perlas Flow-Set Pro. Las estrategias de ventanas de selección empleadas para los tubos del panel, permitieron determinar las poblaciones leucocitarias y subpoblaciones de linfocitos. (Fig. 2). Además, garantizaron la realización de los controles requeridos para la calidad y precisión de los resultados. Los porcentajes de linfocitosen sangre periféricade sujetos sanos, estuvieron dentro de los rangos esperados y en correspondencia con la expresión de tres y cinco moléculas. En las muestras especiales se requieren más estudios que establezcan estos rangos de referencias, a lo que este estudio contribuyó.
Se realizaron controles de eventos positivosy se excluyeron del análisis todos los eventos que no aparecieron adecuadamente en la regióncorrespondiente de los gráficos FS vs SS, CD45 vs SS y de fluorescencia global,en la lógica de ventanas en reversa,. (Fig. 2A y 2B)
El diseño realizado mostró un funcionamiento óptimo en los tres tipos de muestras. Sin embargo, en las muestras de ascitis y tejido los detritus y agregados celulares se solaparon con las poblaciones linfocitarias, esto dificultó la delimitación de las regiones de interés. Se redujeron las gravedades en la centrifuga a 400g, en todos los pasos de centrifugación y se añadió EDTA al 1mM al búfer de lavado. De esta forma disminuyó considerablemente la superposición de detritus y agregados celulares.
Discusión
La CF en el cáncer ha sido muy utilizada para el inmunofenotipo de hemopatías malignas con gran aplicabilidad y su gran aporte al estudio de la enfermedad mínima residual. En la actualidad se ha extendido al estudio de muestras especiales, como distintos líquidos corporales y de efusión, que cumplen el prerrequisito de poseer células en suspensión de forma natural. No obstante, la utilidad de esta tecnología al estudiar tejidos sólidos, ha devenido en necesidad, en cuanto a inmunofenotipo se refiere, por su superioridad en cuanto a sensibilidad, automatización y velocidad de análisis con respecto a la citología e histopatología.15-16
El inmunofenotipado ha evolucionado tanto que ha permitido la identificación de numerosas nuevas subpoblaciones de linfocitos, pero se requieren un promedio de entre 8 y 17 marcajes simultáneos sobre una misma célula.17
El inmunofenotipaje de linfocitos propuestopermitió analizar células circulantes en sangre periférica y líquido ascítico, considerado una extensión del microambiente del tumor de ovario, y el propio tejido ovárico. Este tipo de análisis coincide con los estudios pioneros de Curiel y cols.,18 seguido recientemente, por Jang y cols.19 que proponen evaluaciones simultáneas de subpoblaciones linfocitarias en varios compartimentos del organismo.
Los antígenos seleccionados permitieron caracterizar linfocitos T, B y NK de forma básica y, algunas de sus subpoblaciones de forma multiparamétrica, como NKT, NK secretoras y NK citotóxicas, T doble negativas, T activadas, y de estas últimas, la proporción de Treguñadoras, por un marcaje básico (CD3+, CD4+, CD25alto). Se tuvo en cuenta aquellas que tienen un valor científicamente fundamentado en la inmunopatogenia, evolución, respuesta terapéutica y supervivencia en el cáncer de ovario y que se evalúan en la toma de decisiones clínicas. La combinación de antígenos empleados fue similar a las propuestas en trabajos previamente publicados.5-6,18-20
La determinación porcentual de subpoblaciones linfocitarias en sangre de controles sanos mediante la metodología diseñada, no difiere de los rangos reportadosen la literatura internacional;21) ni de los valores referenciales reportados en estudios cubanos.22-23
La titulación de los conjugados tiene como objetivo disminuir el ruido de fondo por sobresaturación y falsos resultados que esto provoca. De esta forma se asegura la sensibilidad, especificidad y calidad de los resultados. Las ventajas al usar perlas de captura para realizar de forma automática la compensación, incluyen la uniformidad del marcaje, la disminución del error humano y eliminación de la variabilidad de expresión de los antígenos, que ocurre cuando se usan células.No se usaron controles isotípicos, ya que no se recomiendan cuando se usan controles negativos por muestra y controles negativos internos, cuando el marcaje es superficial y uso de las ventanasde selección sobre CD45+.24
Los fluorósforos de tipo tándem se adicionaron al final, ya que la pérdida de su estabilidad, puede interferir en los patrones de expresión y la MIF de otros marcadores, incluso dar resultados falsos positivos en el canal correspondiente alfluorósforoprimario.17
En los diferentes compartimentos del organismo las concentraciones de subpoblaciones linfocitarias varían considerablemente. La metodología propuesta en este estudio permitió determinar esta variabilidad.En este sentido el estudio de Kovacsovics-Bankowski y cols. evidencia, con una metodología similar, diferencias en cuanto a la distribución de linfocitos entre la sangreperiférica, ascitis y tejido tumoral, en pacientes con cáncer de ovario.25
La CF se utiliza actualmente para clasificar los tumores eninmuno-desierto, inmuno-excluido e inmuno-inflamado; teniendo en cuenta el infiltrado de células del sistema inmune. El cáncer de ovario es uno de los tumores más inmunosensibles que existen, generalmente se consideran inmuno-excluidos con predominio supresor. Caracterizar ese infiltrado se ha convertido en una regla de oro en estas pacientes para establecer la conducta terapéutica.26-29
El panel multicolor y la metodología creados permitieron inmunofenotiparlas subpoblaciones linfocitarias por CF, en muestras de sangreperiférica, ascitis y tejido tumoral ovárico; con resultados acorde a los reportados en la literatura especializada, de forma óptima y reproducible. Esta metodología es flexible y permite adicionar nuevos marcadores para analizar otros fenotipos celulares para estudios del microambiente tumoral, así como el inmunofenotipaje en otras enfermedades que posean una inmunopatogenia definida.

