










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión On-line ISSN 1561-3011
Rev Cubana Invest Bioméd vol.34 no.2 Ciudad de la Habana abr.-jun. 2015
Rev Cubana de Investigaciones Biomédicas. 2015;34(2)
ARTÍCULO DE REVISIÓN
El sistema antioxidante del glutatión en la etiopatología de la disfunción nigro-estriatal
Glutathione antioxidant system in the etiopathology of nigrostriatal dysfunction
MSc. Mei-Li Díaz-Hung,I Dra. María Elena González Fraguela,I DrC. Lisette Blanco LezcanoII
I Departamento de Neuroinmunoquímica. Centro Internacional de Restauración Neurológica. La Habana, Cuba.
II Departamento de Neurofisiología Experimental. Centro Internacional de Restauración Neurológica. La Habana, Cuba.
RESUMEN
La enfermedad de Parkinson es una enfermedad neurodegenerativa crónica que afecta a las personas de la tercera edad. En una minoría de los casos la enfermedad es de origen genético pero en el resto, la causa es idiopática. En este sentido, la acumulación de los radicales libres y la pérdida de la homeostasis del glutatión se han señalado como posibles agentes causales. El presente texto se propuso revisar las evidencias experimentales que apoyan la participación de los radicales libres y la pérdida de la homeostasis del glutatión en el comienzo y la progresión de la degeneración de la substantianigrapars compacta. El estrés oxidativo en la enfermedad de Parkinson´s puede estar relacionado con las propiedades pro-oxidantes intrínsecas de la dopamina y elevadas concentraciones de hierro en la substantianigrapars compacta, que promueven la oxidación de la dopamina y la generación de especies reactivas del oxígeno. Cualquier evento que desencadene estos mecanismos, genera un daño celular. La disminución del glutatión es una de las alteraciones bioquímicas más tempranas, detectadas en asociación con la enfermedad de Parkinson y se ha relacionado con la inhibición del complejo I de la cadena de transporte mitocondrial, daño oxidativo, activación glial, entre otros que favorecen la neurodegeneración. Estas evidencias sugieren la necesidad de mantener la homeostasis del glutatión en el sistema dopaminérgico y su vínculo con la etiología de la degeneración nigro-estriatal, lo que tiene una potencial aplicación en la práctica clínica.
Palabras clave: radicales libres, estrés oxidativo, glutatión, ganglios basales, sistema dopaminérgico, neurodegeneración.
ABSTRACT
Parkinson’s disease is a chronic neurodegenerative condition affecting elderly persons. In a minority of cases the disease has a genetic origin, but in most the cause is idiopathic. Accumulation of free radicals and loss of glutathione homeostasis have been pointed at as possible causal agents. The purpose of the study was to review experimental evidence supporting the involvement of free radicals and loss of glutathione homeostasis in the outset and progress of substantia nigra pars compacta degeneration. Oxidative stress in Parkinson’s disease may be related to the intrinsic pro-oxidant properties of dopamine and high iron concentrations in the substantia nigra pars compacta, promoting dopamine oxidation and the generation of reactive oxygen species. Any event triggering these mechanisms will cause cell damage. Glutathione reduction is one of the earliest biochemical alterations detected in association with Parkinson’s disease, and it has been related to the inhibition of complex I of the mitochondrial transport chain, oxidative damage and glial activation, among other factors leading to neurodegeneration. This evidence points to the need to maintain glutathione homeostasis in the dopaminergic system, as well as its relationship to the etiology of nigrostriatal degeneration, of potential application in clinical practice.
Key words: free radicals, oxidative stress, glutathione, basal ganglia, dopaminergic system, neurodegeneration.
INTRODUCCIÓN
El estrés oxidativo es un fenómeno dinámico y complejo caracterizado por un desbalance entre la generación de especies reactivas de oxígeno (ERO) y la disponibilidad y acción de los antioxidantes.1 El tejido nervioso está formado por células post-mitóticas y además, tiene alto consumo de oxígeno, contenido de lípidos y actividad metabólica que hacen que sea en particular, susceptible al daño oxidativo. Durante el envejecimiento o en condiciones patológicas, la oxidación de las biomoléculas se incrementa y aumentan las cantidades de proteínas, lípidos y ácido desoxirribonucleico (ADN) oxidados.2
La eliminación rápida de ERO ocurre por la combinación de enzimas antioxidantes con una alta actividad catalítica.3 Además, existen compuestos antioxidantes de bajo peso molecular como el β-caroteno, el ácido retinoico, el α-tocoferol, el ácido ascórbico, la coenzima Q, quelantes del hierro como la ferritina, la melatonina, el ácido úrico y tioles como el glutatión (GSH) que neutralizan espontáneo las ERO y sus productos de reacción.4
El GSH fue descubierto en 1888 por Rey Pailhade en la levadura pastelera, su estructura fue identificada en 1930 y a partir de 1970 un considerable número de trabajos demostró su importancia en la biología celular.5 El GSH reacciona directo con las ERO y actúa como cofactor de enzimas antioxidantes como la GPx. Además, mantiene el potencial redox celular al preservar en estado reducido los grupos sulfidrilos de las proteínas y regula la señalización celular de la apoptosis.6 Las alteraciones en la síntesis del GSH o en su contenido se han asociado con una variedad de enfermedades neurodegenerativas como la esquizofrenia, la enfermedad de Alzheimer y la enfermedad de Parkinson (EP).7
La EP es una enfermedad crónica neurodegenerativa que afecta un aproximado de 9,5/1000 personas mayores de 65 años. Los síntomas cardinales de la enfermedad incluyen bradicinesia, rigidez muscular, temblor en reposo e inestabilidad postural.8 Algunos síntomas no motores preceden el desarrollo de los síntomas motores clásicos como constipación, trastornos del sueño REM (del inglés rapideyesmovement), depresión, hiposmia, apatía, reacción lenta y dolor. Estos síntomas están asociados a la disfunción de estructuras no dopaminérgicas del Sistema Nervioso Central (SNC). Se ha propuesto que la patología de EP comienza en el tronco encefálico y estructuras olfatorias y progresa siguiendo un patrón rostro-caudal.9
Aunque múltiples sistemas neuronales estén implicados, la característica neuropatológica distintiva de la EP es la degeneración de la substantianigrapars compacta (SNpc).10 Esta área es el origen de las proyecciones dopaminérgicas al complejo caudado/putamen y su degeneración, causa la disfunción del circuito de los núcleos basales.11 La enfermedad es esporádica, aunque en una minoría de los casos está relacionada con mutaciones familiares. La etiología de la enfermedad es desconocida pero el estrés oxidativo y la disfunción mitocondrial parecen desempeñar un papel crucial en la muerte de las neuronas dopaminérgicas.12 El objetivo de este trabajo es revisar las evidencias experimentales que apoyan la participación de los radicales libres y la pérdida de la homeostasis del GSH en el comienzo y la progresión de la degeneración de SNpc.
EL CONTROL REDOX Y SU RELACIÓN CON LAS ALTERACIONES MOTORAS DE LA ENFERMEDAD DE PARKINSON
Radicales libres, estrés oxidativo y defensas antioxidantes
El término “radicales libres” designa una familia de compuestos caracterizados por su gran reactividad debido a que poseen un electrón desapareado en el orbital externo. A este grupo pertenecen tanto las ERO, que incluyen el superóxido (O2-), el hidroxilo (OH-), el hidroperóxido (HO2-) y el peróxido de hidrógeno (H2O 2), como las especies reactivas del nitrógeno (ERN), tales como el óxido nítrico (NO) y el peroxinitrito (ONOO-). La fuente endógena de ERO más importante es el sistema mitocondrial de transporte de electrones (Fig. 1) que utiliza casi el 90 % del O 2 consumido para la fosforilación oxidativa. Debido a este elevado flujo, es inevitable el escape de electrones, que resulta en la formación de O2- fundamental, en el complejo I (NADH deshidrogenasa) y el complejo III (semiubiquinona).13
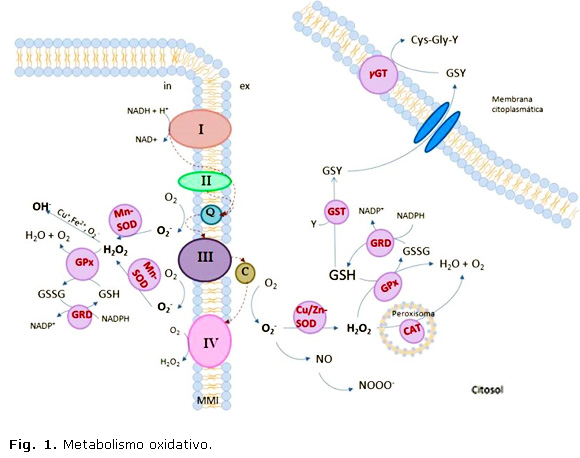
El anión O2- se reduce espontáneo a H2O2. El H2O2 no es un radical libre pues no presenta electrones sin parear, pero es estable en solución y puede atravesar las membranas libres o a través de canales acuaporinas y así difundir a un compartimiento diferente al que le dio origen. El anión O2- interactúa con el H2O2 (reacción de Haber-Weiss) y forma OH- y con el NOforma ONOO-. Otra vía de generación del OH - transcurre mediante la reacción de Fenton en la que tiene lugar la reducción de H2O2 por iones metálicos como el hierro (Fe2+) y el cobre (Cu+).14
Debido a que los organismos poseen mecanismos de defensa antioxidantes, el estrés oxidativo ha sido definido como “un exceso de producción de sustancias pro-oxidantes, un déficit de los mecanismos de defensa contra la oxidación, o a ambos factores”.15 Este concepto ha sido útil para la investigación desde 1985 cuando fue formulado por Helmut Sies. Sin embargo, la acumulación de datos experimentales en las vías de señalización redox, los ensayos de intervención con antioxidantes y los marcadores de estrés oxidativo sugieren que una definición más adecuada es “una ruptura de la señalización y el control redox”.16
Numerosas evidencias indican que las ERO y las ERN actúan como moléculas de señalización y modulan una variedad de funciones celulares como la transcripción de genes, la traducción, el metabolismo, la proliferación y la apoptosis.17 Las cascadas de señalización en las que participan, incluyen la activación de receptores tirosina quinasa, fosforilación- desfosforilación de proteínas, cierre (disminución de la probabilidad de apertura) de canales KATP, la cascada del fosfatidilinositol-3-quinasa y la activación de proteína G. A estos eventos subyacen la activación de las MAPK (del inglés mitogen-activatedproteinkinase) y factores de transcripción como el factor nuclear NF-κB y el activador de proteína 1 (AP1).18
En elevadas cantidades, las ERO son tóxicas debido a su reacción con los lípidos, las proteínas y los ácidos nucleicos. La correcta respuesta a la producción de ERO es crítica para prevenir el daño oxidativo y para mantener la supervivencia celular. El daño a los componentes celulares desencadena la muerte celular por procesos tanto de apoptosis como de necrosis, depende de la magnitud del estrés oxidativo.19 Entre las enzimas con función antioxidante, la superóxido dismutasa cataliza la conversión del O2 - en H2O2 el cual es degradado por enzimas como la catalasa20 y la glutatión peroxidasa (GPx). Esta última es la principal enzima que degrada los peróxidos en el sistema nervioso y cataliza con alta especificidad la descomposición del H 2O2 y de peróxidos orgánicos, utiliza el GSH como donador de electrones.6 La glutatión reductasa (GRD) cataliza la reducción del glutatión oxidado (GSSG), lo que mantiene el nivel de GSH en la célula. Por último, la glutatión- S-transferasa (GST) conjuga el GSH con muchos compuestos orgánicos; puede reducir hidroperóxidos y también detoxifica el hidroxinonenal (HNA), un producto de la peroxidación lipídica.19
Homeostasis y funciones del glutatión
El tripéptido GSH (γ-L-glutamil-L-cisteína-glicina) es un tiol presente en concentraciones de hasta 12mM en las células de los mamíferos. Se sintetiza in vivo por la acción consecutiva de dos enzimas que utilizan adenina-3-fosfato (ATP) (Fig. 2). La γ-glutamil-cisteína sintetasa (GCS) utiliza glutamato y cisteína para formar el dipéptido γ-glutamil-cisteína que al final se combina con glicina en una reacción catalizada por la glutatión sintetasa.21
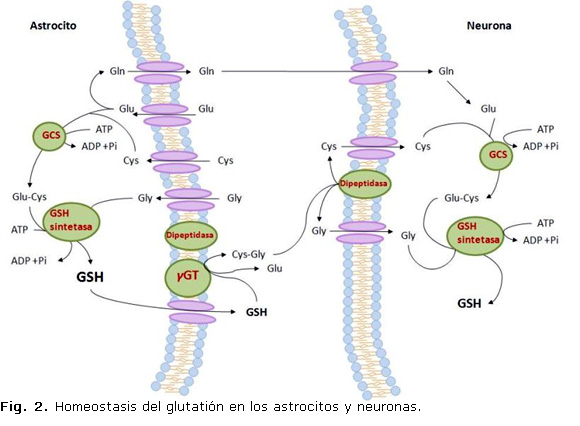
A pesar de ser sintetizado exclusivo en el citosol, el GSH se encuentra distribuido en el retículo endoplasmático, el núcleo y la mitocondria. En cada uno de ellos la relación GSH/GSSG es diferente, lo que garantiza su correcto funcionamiento. En el núcleo, el GSH mantiene en su forma reducida a los grupos sulfidrilos de las proteínas necesarios para la reparación del ADN. En el retículo endoplasmático, se encuentra de manera predominante la forma GSSG, lo que permite la formación de puentes disulfuro y el correcto plegamiento de las proteínas durante su síntesis. En la mitocondria, la concentración de GSH es similar a la citosólica, lo cual es de gran importancia ya que este organelo está expuesto directo a las ERO.22
En el SNC tiene lugar una interacción entre los astrocitos y las neuronas durante la síntesis de GSH que es de gran importancia para proteger a las neuronas del ataque de las ERO.23 Los astrocitos tienen mayor concentración de GSH y mayor capacidad de secretar GSH al espacio extracelular que las neuronas. Esta secreción sirve para suplir de GSH a otras células del cerebro, incluyen las neuronas, mediante la γGT. La síntesis de GSH depende de la disponibilidad de la cisteína. La incorporación de este aminoácido ocurre en las neuronas a través de un transportador para aminoácidos excitatorios (EAATs) mientras que en los astrocitos ocurre a través del intercambiador cistina/glutamato (Xc-).24
El nivel de GSH es regulado fundamental por el control de su síntesis y exportación de las células y puede afectarse por agentes o condiciones que alteren el estado redox o por ruptura de la distribución entre los organelos.25 El GSSG obtenido durante el curso de la reacción de la GPx es reciclado a GSH por la GRD, de manera que la relación GSH/GSSG sirve como indicador del ambiente redox celular. 16 En contraste, el GSH reacciona espontánea con el O2- el NO, el OH- y el ONOO-6 y es consumido durante la generación de glutatión-S-conjugados por la GST en la eliminación de xenobióticos o compuestos endógenos, o por su liberación de la célula. Estos procesos disminuyen el nivel de GSH total y mantienen su concentración intracelular, cuando el GSH consumido es remplazado por resíntesis a partir de sus aminoácidos precursores. El GSH extracelular y sus conjugados son sustrato de la enzima γ-glutamil-transpeptidasa (γGT).
El GSH también participa en la regulación del transporte de aminoácidos hacia la célula, en el mantenimiento de la estructura tridimensional de las proteínas y en la señalización redox.26 Además, el GSH sirve como transporte/almacén de cisteína, la cual por sí misma tiene efectos neurotóxicos.27 Como neuromodulador/neurotransmisor, el GSH se une los receptores NMDA y exhibe una acción dual (agonista/antagonista) en las repuestas neuronales mediadas por estos receptores. También sirve como reservorio endógeno de NO al formar S-nitroso glutatión que puede liberar el NO en ciertas condiciones y tiene un efecto protector en el cerebro en condiciones de estrés oxidativo. Además, el GSH se requiere para la proliferación celular y la diferenciación neuronal.5
Núcleos grises de la base y su relación con el sistema motor
Los núcleos basales (ganglios basales) consisten en cuatro núcleos:
1. El estriado (EST) subdividido en los núcleos caudados y putamen.
2. El globo pálido con sus segmentos externo e interno (GPe y GPi al respecto).
3. El núcleo subtalámico (NST).
4. La sustantianigra en sus porciones reticulada (SNpr) y compacta (SNpc).
Ellos están involucrados en circuitos segregados anatómico que funcionan en paralelo: “motor”, “oculomotor”, “asociativo” y “límbico”.28 Cada uno de estos circuitos recibe aferencia de regiones corticales funcional relacionadas. Por ejemplo, el circuito motor recibe de las áreas: motora, premotora, motora suplementaria y somatosensorial. La eferencia de los ganglios basales es hacia una única región frontal; en el caso del circuito motor, hacia el área motora suplementaria.29 El circuito motor está involucrado en el control del movimiento y la disfunción en elementos de estos circuitos se relaciona con trastornos del movimiento como la EP.30
La actividad de las neuronas de los núcleos basales, tributa a diferentes parámetros del “plan motor” elaborado en las áreas motoras de la corteza como dirección, velocidad, amplitud, carga y fuerza muscular. Estos núcleos están relacionados no con un único movimiento, sino más bien con combinaciones de ellos y presentan una fina organización somatotópica.31 En este sentido, el GPi está involucrado en el control del movimiento de la musculatura axial y de las extremidades, mientras que la SNpr participa en el control motor de la cabeza y los ojos.32
En el modelo clásico de los núcleos basales (Fig. 3) desarrollado en la década de 1980 por Albin, Young y Penney33 el EST (GABAérgico) recibe aferencias glutamatérgicas procedentes de áreas específicas de la corteza y del tálamo, y transfiere la información a los núcleos de salida, el GPi y la SNpr (GABAérgicos). De acuerdo a este modelo, las proyecciones entre el EST y el GPi/SNpr se dividen en dos vías separadas. Una “vía directa” (monosináptica) y una “vía indirecta” a través del GPe (GABAérgica) y el NST (glutamatérgica). La eferencia del GPi/SNr se dirige hacia los núcleos ventrolateral y ventral anterior del tálamo, el cual proyecta de regreso hacia la corteza cerebral.32 A la vez el estriado también recibe una proyección dopaminérgica, que se origina en la SNpc (vía nigro-estriatal) y modula la transmisión corticoestriatal.28

La “vía directa” se origina en las neuronas espinosas de talla mediana (NEM) del EST que expresan receptores D1, sustancia P y dinorfina y produce inhibición de las neuronas del GPi/SNpr. La inhibición de los núcleos GPi/SNpr desinhibe los núcleos talámicos diana, lo que activa las proyecciones talámicas a la corteza y por tanto la función motora. Esta vía es facilitada por la transmisión dopaminérgica mediante los receptores D1.32
La “vía indirecta” se origina en las NEM que expresan receptores D2 y encefalinas y proyecta hacia el GPi/SNpr a través del GPe y el NST. En este caso, las NEM inhiben el GPe, esta inhibición “libera” el NST que puede entonces estimular el GPi/SNpr. La actividad del GPi/SNpr mantiene a los núcleos motores del tálamo y al tallo cerebral bajo un control inhibitorio que impide o detiene el movimiento. La acción de la DA inhibe la “vía indirecta” a través de los receptores D2.33 Otras estructuras como el núcleo pedúnculo pontino (NPP) parecen proveer una interface entre el movimiento y las estructuras del cerebro anterior implicadas en la atención a los estímulos sensoriales y la actividad motivacional.34
En la actualidad se considera que los núcleos basales no solo son codificadores de las habilidades motoras sino que también están involucrados en el aprendizaje de tales habilidades. La actividad recurrente y alternante de las neuronas de los núcleos basales participa en los procesos relacionados con el aprendizaje motor. Esto ocurre mediante sincronización de la actividad y composición de secuencias entre diferentes microcircuitos neurales.35
Recién el NST se ha considerado como otra estación de entrada que recibe aferencia directa desde la corteza (“vía hiperdirecta”), el tálamo y el tallo cerebral. Junto a las proyecciones eferentes del NST conocidas, se ha confirmado la presencia de proyecciones directas del NST al tálamo ventral. También se han reconocido conexiones recíprocas entre el GPe y el EST, y entre el EST y la SNpc. El NST-GPe-GPiforman un microcircuito donde el GPe aparenta estar posicionado para controlar la actividad eferente de los núcleos basales. Asimismo, el sistema de proyección dopaminérgico inerva además del EST, estructuras como el NST, el GPe y el GPi, así como áreas de corteza, núcleos del tálamo y estructuras del sistema límbico.33
Las vías “directa” e “indirecta” son coactivadas durante la iniciación del movimiento y se inactivan cuando el animal está en reposo. Además, la activación de ambas vías precede el inicio del movimiento.11 Se plantea que cada acción motora posible, está representada por un pequeño grupo de neuronas en las estructuras eferentes de los núcleos basales. La inhibición selectiva de un grupo facilita las neuronas corticales que codifican la acción motora seleccionada. Al mismo tiempo, la facilitación de otros grupos neuronales en el GPi/SNpr inhibe otras acciones motoras que compiten. De este modo, la “vía directa" desde el EST genera focos de inhibición en subpoblaciones neuronales del GPi/SNpr mientras que las proyecciones a través del NST y el GPe (por las vía hiperdirecta e indirecta) exhibe excitación difusa del GPi/SNpr. El efecto combinado genera el patrón de activación.31
Sistemas dopaminérgicos del cerebro medio. Receptores dopaminérgicos
Existen tres sistemas dopaminérgicos fundamentales en el cerebro. Los cuerpos celulares de la vía nigroestriatal residen en la SNpc y proyectan hacia el EST dorsal (caudado-putamen); la degeneración de esta vía resulta en el trastorno motor de la EP. La vía mesolímbica se origina en el área tegmental ventral y termina en el núcleo acumbens; una función de este sistema es la mediación de la satisfacción natural e inducida por drogas. La vía mesocortical también se origina en el área tegmental ventral y alcanza la corteza prefrontal, regula complejos procesos cognitivos como la atención selectiva y la memoria de trabajo.36
La liberación de DA tanto en el cerebro medio como en el cerebro anterior, es regulada dinámica y local por el microcircuito que rodea el sitio de liberación. Los datos indican que la señal dopaminérgica puede ser inhibida o potenciada de manera independiente a la frecuencia por una variedad de factores que regulan la liberación axonal o somatodendrítica. Estos factores incluyen la recaptura de la DA mediada por el transportador, localizado en la neurona dopaminérgica, los autorreceptores, el ion Ca++, los neurotransmisores glutamato, GABA y acetilcolina, opiodes, cannabinoides, el H2O2 y el NO.37
La acción de la DA en su sitio blanco es mediada por una familia de receptores acoplados a proteína G codificados por al menos 5 genes (D 1, D2, D3, D4 y D5). Estos receptores están clasificados en dos subfamilias, los receptores tipo D1 y los receptores tipo D2 basado en la homología de secuencia y propiedades farmacológicas.38 Los receptores de tipo D1 (D1 y D5), están acoplados a proteína Gαs que estimula la adenilato cliclasa e incrementa el nivel de AMPc con la subsiguiente activación de proteína quinasa A (PKA) y quinasas dependientes de señales extracelulares. Los receptores de tipo D2 (D2, D3 y D4) están de ningún modo acoplados a esta señalización dependiente de AMPc/PKA.39 La activación de estos receptores también modula los niveles de Ca++ intracelular por la estimulación de la hidrólisis del fosfatidil inositol por la fosfolipasa C a inositol-3-fosfato (IP3), el cual moviliza el calcio de los reservorios intracelulares.40
La modulación dopaminérgica depende del subtipo del receptor, su localización en el sitio pre o postsináptico, la concentración de DA en el microambiente y el estado de actividad de la neurona blanco. En las NEM estriado convergen los axones de las neuronas dopaminérgicas de la vía nigroestriatal y las proyecciones glutamatérgicas corticoestriatales. Así, la activación del receptor D1 facilita la transmisión corticoestriatal a través, de la “vía directa” mientras que la activación de los receptores D2 inhibe la transmisión a través de la “vía indirecta”.41 Además, la DA liberada en el EST está implicada en los procesos de aprendizaje y plasticidad sináptica tales como la depresión (LTD, del inglés longtermdepression) o potenciación (LTP, del inglés longtermpotentiation) a largo plazo.30
La DA también tiene acción sobre otras estructuras de los núcleos basales, además, del EST (Fig. 3). Los autoreceptores D2 suprimen la liberación somatodendrítica de DA en la SNpc y la liberación axonal en el EST.42 En el GPe, la DA inhibe la transmisión GABAérgica procedente de las NEM de la “vía indirecta” y estimula la liberación de Glu procedente del NST, lo que resulta en un incremento de la actividad del GPe. La DA actúa sobre receptores D2 localizados en las terminales presinápticas e inhibe la liberación de GABA en el NST, facilita su actividad. En el GPi, actúa sobre los receptores tipo D1 en las terminales presinápticas y facilita la liberación de GABA.30
Bases neurales de las alteraciones motoras asociados a las neuronas dopaminérgicas de la SNpc. Factores patogénicos
El EST y los núcleos basales han sido implicados en una amplia variedad de trastornos psicomotores como la EP, la esquizofrenia y el abuso de drogas. La alteración de la regulación de las vías “directa” e “indirecta” por la DA parece ser un aspecto esencial en la mayoría de estas enfermedades. El ejemplo mejor caracterizado es la EP, en la que las neuronas dopaminérgicas de la SNpc degeneran. Los estudios en modelos experimentales de EP sugieren que la excitabilidad de las vías “directa” e “indirecta” cambia luego de la disminución de los niveles de DA, y crea un desbalance en la regulación de los núcleos motores del tálamo que favorece la supresión del movimiento.32
En el EST, blanco principal de la inervación dopaminérgica, ocurren una serie de cambios celulares y sinápticos en repuesta a la deficiencia de DA. Se observa un incremento compensatorio en la respuesta de los receptores D1 y D2, así como un incremento en la actividad de las neuronas involucradas en la “vía indirecta” del circuito motor.43 La pérdida de la señalización mediada por los receptores D1 y D2 promueve los mecanismos de LTD en la “vía directa” y de LTP en la “vía indirecta”, al respecto. De modo que los cambios en la fortaleza sináptica dependientes de la actividad ocurren en paralelo, a los cambios de excitabilidad que siguen a la disminución de la DA.32
El efecto neto de este desbalance entre las vía “directa” e “indirecta” es una elevada actividad de las neuronas inhibitorias del GPi que proyectan al tálamo. La inhibición de este centro premotor subyace a los síntomas de acinesia y bradicinecia de la EP, aunque no explican completo el temblor, la rigidez y los síntomas axiales.44 Existen evidencias de actividad sincrónica en la frecuencia de oscilación entre el NST, el GPe y la corteza motora. Esto podría conducir a una ruptura de la capacidad de las neuronas individuales de procesar y transmitir información específica y de este modo, controlar movimientos complejos. La generación de este tipo de actividad no está claramente comprendida, aunque es posible que la vía hiperdirecta esté involucrada y esté promovida por los cambios en las propiedades sinápticas producidos por la pérdida de la DA.45
Además, de la degeneración de las neuronas de la SNpc, es característico de la EP la presencia de cuerpos de inclusión citoplasmáticos eosinófilos (cuerpos de Lewy) formados en lo principal por α-sinucleína fibrilar y ubiquitina. Estos están presentes en la SNpc y otras estructuras como el NPP, el bulbo olfatorio, núcleo rafe, locus coeruleos y neuronas post-ganglionares del Sistema Nervioso Autónomo. Estas lesiones contribuyen a los síntomas no motores de la enfermedad que en lo general aparecen temprano en el curso de la misma. Los síntomas motores aparecen cuando el nivel de DA es inferior al 60 % de lo normal.46
Los mecanismos involucrados en la neurodegeneración continúan poco dilucidados, sobre todo porque la mayoría de los casos de EP son de causa desconocida. Las evidencias actuales apoyan la disfunción mitocondrial, el estrés oxidativo y la acumulación de proteínas mal plegadas como factores desencadenantes de la muerte neuronal. Otros mecanismos que pueden contribuir al proceso degenerativo son la neuroinflamación y la excitotoxicidad, así como aspectos metabólicos y funcionales específicos de las células dopaminérgicas.47
En la SNpc de pacientes de EP se ha observado un daño oxidativo en el ADN y en las proteínas, así como un incremento del producto más importante de la oxidación lipídica, el 4-hidroxi-2-nonenal (HNE).48 Además, la actividad de la enzima monoamino-oxidasa (MAO) está aumentada en la EP lo que implica estimulación del metabolismo dopaminérgico por la acción de esta enzima y una formación excesiva de H2O2. Por otro lado, la actividad de la enzima óxido nítrico sintasa se encuentra incrementada, estimula la producción de óxido nítrico (NO) que se combina con el radical superóxido para formar peroxinitrito (ONOO-), lo que exacerba el estrés oxidativo y la disfunción mitocondrial.49 La disipación del potencial de membrana de la mitocondria y la liberación de citocromo C en el citoplasma pueden activar las cascadas apoptóticas.46
El estrés oxidativo en la EP puede estar relacionado con las propiedades pro-oxidantes intrínsecas de la dopamina (DA) y elevadas concentraciones de hierro en la SNpc que promueve la oxidación de la DA y la generación de ERO.47 Aunque se almacena en vesículas, el exceso de DA citosólica se oxida con facilidad de forma espontánea y enzimáticamente produciendo quinonas. Estas especies son capaces de modificar diversas proteínas y pueden conducir a la inactivación de:
- El trasportador de DA (DAT).
- La enzima tirosina hidroxilasa (TH).
- La cadena de transporte mitocondrial.
La degradación de la DA por la MAO genera H2O2 que puede ser convertido rápido a OH- en presencia de metales de transición.50 La neuromelanina es otro producto de la oxidación de la DA que tiene la capacidad de almacenar y liberar grandes cantidades de hierro y puede inducir la activación de la microglia.12
Debido a que las neuronas dopaminérgicas son generadoras de ERO y en lo especial vulnerables al estrés oxidativo, cualquier evento que desencadene estos mecanismos puede dañar a la célula.50 La disminución del GSH es una de las alteraciones bioquímicas más tempranas detectadas en asociación con la EP, como demostró la observación de que la pérdida de GSH ocurre en la enfermedad de cuerpos de Lewy incidental, considerada como un antecedente asintomático de la EP.51
La neuroinflamación es otro de los mecanismos que puede participar en la degeneración dopaminérgica nigral. Estas células expresan receptores a algunas citoquinas como el factor de necrosis tumoral (TNF-α), la interleucina 1β y el interferón γ las cuales pueden dañar las células dopaminérgicas, que inducen enzimas como la óxido nítrico sintasa, la ciclo-oxigenasa y la NADPH oxidasa que generan especies tóxicas. Además, las células dañadas pueden liberar productos que activan la microglia y perpetúan la degeneración. Otro factor que contribuye a la patogénesis es la excitotoxicidad cuando se exacerba la señalización de glutamato proveniente del NPP y el NST.52
La homeostasis del glutatión y los mecanismos de degeneración nigral
Aunque el GSH no es la única molécula antioxidante que se ha reportado alterada en la EP, la magnitud de su disminución está en correspondencia con la severidad de la enfermedad y es el indicador más temprano de degeneración nigro-estriatal. La disminución del GSH conduce a un aumento en la producción de NO y la recaptura de hierro en células dopaminérgicasin vitro.53 Además, se ha demostrado la inhibición del complejo I de la cadena de transporte mitocondrial, luego del tratamiento con L-butioninsulfoximina (BSO), un inhibidor de la GCS, enzima que limita la síntesis de GSH.
La disminución del GSH puede resultar en una significativa inhibición de la actividad del complejo I mitocondrial mediada por NO, así como en una reducción de la actividad del complejo II y de la producción de ATP, lo que deviene en un aumento en la generación de ERO. 54 Entre todas las disfunciones mitocondriales descritas en las enfermedades neurodegenerativas, la deficiencia del complejo I parece ser en lo relativo, específica de la EP.55
Los estudios acerca de la deficiencia de GSH como factor etiológico en la degeneración de la SNpc muestran resultados heterogéneos. Se plantea que la disminución del GSH por sí sola no conlleva la muerte de las células dopaminérgicas.56 Sin embargo, en cultivo mixto de neuronas y células gliales bajo tratamiento con BSO, se observó una disminución del número de células positivas a la TH, un incremento de las células en apoptosis y activación glial, también observada en muestras de SNpc de pacientes parkinsonianos. 57 Posterior, Garrido y colaboradores encontraron degeneración de la SNpc y moderada astrogliosis debido a la inhibición de la síntesis del GSH. Sorprendente, estos autores mostraron que la sobreexpresión de la GCS también puede iniciar la degeneración de las células dopaminérgicas, lo que sugiere que el metabolismo del GSH debe ser controlado.58
Los astrocitos pueden proteger a las neuronas de la toxicidad inducida por las ERO y son en lo exclusivo importantes para la detoxificación del H 2O2, ya que aportan cisteína a las neuronas para la síntesis de GSH.23 Además, expresan el factor Nrf-2, el cual, en respuesta al estrés oxidativo promueve la transcripción de proteínas antioxidantes como la NADPH quinona oxidoreductasa, la GST y las subunidades moduladoras y catalíticas de la GCS. El Nrf-2 potencia de esta forma la síntesis de GSH en los astrocitos, y su sobre expresión conduce a neuroprotección en modelos in vivo de EP.59
A pesar de que la activación glial es beneficiosa para las neuronas, la activación continua o repetida de los astrocitos y la microglia puede conducir al incremento de la producción de radicales libres y otros mediadores neurotóxicos, que puede conducir a daño neuronal. 60 La activación glial combinada con la disminución del GSH, lo cual incrementa la actividad lipooxigenasa, resulta en un exceso de generación de radicales libres y crea un reto oxidativo adicional para las células con bajo contenido de GSH.
Según se plantea, cuando el GSH disminuye el H2O2 producido por la MAO, potencia la liberación de ácido araquidónico mediante la activación de la fosfolipasa A2 de membrana y también puede ser convertido a OH- en presencia de metales como el hierro y el cobre. Los ácidos araquidónico, linoleico y linolénico potencian la formación de hidroperóxidos lipídicos.61 La disminución del GSH también está relacionada con un incremento en la recaptura del hierro en cultivo de células dopaminérgicas a partir de la traducción de la proteína transportadora. Este incremento se acompaña de un aumento en los niveles de radicales libres y es dependiente de H2O2.53
Sin embargo, el incremento de las ERO puede generar una respuesta compensatoria en las enzimas antioxidantes, especial en la CAT como fue mostrado reciente en estudios del laboratorio.62 Se reportó que en la SNpc la actividad enzimática de esta enzima es mayor que la actividad de la GPx.63 El incremento de la actividad de la CAT previene el daño oxidativo a las diferentes biomoléculas y con ello el aumento de lipoperóxidos y otros compuestos electrofílicos como aldehídos y quinonas.64 No obstante, previo, se mostró en ratas tratadas con 6-OHDA que la disminución de DA en la SNpc no está relacionada con una acumulación de los productos de peroxidación lipídica65 lo que sugiere que la disminución de glutatión podría conducir a la muerte celular por un mecanismo independiente del daño oxidativo.
El papel del GSH en la apoptosis no está bien establecido y depende en gran medida del tipo celular. Se ha encontrado que el desbalance GSH/GSSG precede la pérdida de la integridad mitocondrial, la liberación de citocromo C y activación de las caspasas. Estos eventos ocurren en corto tiempo y la recuperación del GSH no impide la apoptosis, lo que indica que la modificación de la señalización redox ocurre de manera temprana.66
La disminución del GSH puede activar las quinasas reguladas por señales extracelulares (ERK2, de sus siglas en inglés). La activación de las MAPK, entre ellas las ERK subyace a la respuesta antioxidante. Su activación se manifiesta en la activación de factores de transcripción como el Nrf-2 y el NF-κB.67 La activación de las ERK puede tener un papel dual en la supervivencia y muerte celular y parece ser un factor crítico en la muerte neuronal inducida por la disminución del GSH.68
Se concluye que la disminución del contenido de GSH puede generar un desbalance oxidativo a favor de la acumulación de ERO y ERN, provocan daño oxidativo a las biomoléculas. Este desbalance puede generar una respuesta compensatoria antioxidante, encaminado a garantizar la sobrevivencia celular. Sin embargo, las ERO/ERN pueden activar cascadas de señalización relacionadas con la muerte celular y activar la glía. La respuesta glial, en dependencia de su magnitud, también puede exacerbar el daño celular y contribuir a la degeneración nigro-estriatal (Fig. 4). Futuras investigaciones en este sentido son necesarias pues el conocimiento de estos mecanismos puede ser de gran utilidad para el diagnóstico y tratamiento temprano de la EP.

REFERENCIAS BIBLIOGRÁFICAS
1. Du ZX, Zhang HY, Meng X, Guan Y, Wang HQ. Role of oxidative stress and intracellular glutathione in the sensitivity to apoptosis induced by proteasome inhibitor in thyroid cancer cells. BMC Cancer. 2009 Feb 16;9:56. doi: 10.1186/1471-2407-9-56.:56-9.
2. Kumar H, Lim HW, More SV, Kim BW, Koppula S, Kim IS, et al. The Role of Free Radicals in the Aging Brain and Parkinson's Disease: Convergence and Parallelism. Int J Mol Sci. 2012 Aug 21;13(8):10478-504.
3. Brandes N, Schmitt S, Jakob U. Thiol-based redox switches in eukaryotic proteins. Antioxid Redox Signal. 2009 May;11(5):997-1014.
4. Brambilla D, Mancuso C, Scuderi MR, Bosco P, Cantarella G, Lempereur L, et al. The role of antioxidant supplement in immune system, neoplastic, and neurodegenerative disorders: a point of view for an assessment of the risk/benefit profile. Nutr J. 2008 Sep 30;7:29. doi: 10.1186/1475-2891-7-29.:29-7.
5. Aoyama K, Watabe M, Nakaki T. Regulation of neuronal glutathione synthesis. J Pharmacol Sci. 2008 Nov;108(3):227-38.
6. Chen J, Small-Howard A, Yin A, Berry MJ. The responses of Ht22 cells to oxidative stress induced by buthionine sulfoximine (BSO). BMC Neurosci. 2005 Feb 12;6(10):10.
7. Chi L, Ke Y, Luo C, Gozal D, Liu R. Depletion of reduced glutathione enhances motor neuron degeneration in vitro and in vivo. Neuroscience. 2007 Feb 9;144(3):991-1003.
8. Blonder LX, Slevin JT. Emotional dysfunction in Parkinson's disease. Behav Neurol. 2011;24(3):201-17.
9. Tolosa E, Pont-Sunyer C. Progress in defining the premotor phase of Parkinson's disease. J Neurol Sci. 2011 Nov 15;310(1-2):4-8.
10. Boll MC, Alcaraz-Zubeldia M, Rios C. Medical management of Parkinson's disease: focus on neuroprotection. Curr Neuropharmacol. 2011 Jun;9(2):350-9.
11. Carvalho MM, Campos FL, Coimbra B, Pego JM, Rodrigues C, Lima R, et al. Behavioral characterization of the 6-hydroxidopamine model of Parkinson's disease and pharmacological rescuing of non-motor deficits. Mol Neurodegener. 2013 Apr 26;8:14. doi:10.1186/1750-1326-8-14.:14-8.
12. Michel PP, Ruberg M, Hirsch E. Dopaminergic neurons reduced to silence by oxidative stress: an early step in the death cascade in Parkinson's disease? Sci STKE. 2006 Apr 25;2006(332):e19.
13. Pervaiz S, Taneja R, Ghaffari S. Oxidative stress regulation of stem and progenitor cells. Antioxid Redox Signal. 2009 Nov;11(11):2777-89.
14. Nita DA, Nita V, Spulber S, Moldovan M, Popa DP, Zagrean AM, et al. Oxidative damage following cerebral ischemia depends on reperfusion - a biochemical study in rat. J Cell Mol Med. 2001 Apr;5(2):163-70.
15. Jimenez-Jimenez FJ, Alonso-Navarro H, Ayuso-Peralta L, Jabbour-Wadih T. Oxidative stress and Alzheimer's disease. Rev Neurol. 2006 Apr 1;42(7):419-27.
16. Jones DP. Redefining oxidative stress. Antioxid Redox Signal. 2006 Sep;8(9-10):1865-79.
17. Palumaa P. Biological redox switches. Antioxid Redox Signal. 2009 May;11(5):981-3.
18. Fisher AB. Redox signaling across cell membranes. Antioxid Redox Signal. 2009 Jun;11(6):1349-56.
19. Morgan MJ, Liu ZG. Crosstalk of reactive oxygen species and NF-kappaB signaling. Cell Res. 2011 Jan;21(1):103-15.
20. Cardenas-Rodriguez N, Huerta-Gertrudis B, Rivera-Espinosa L, Montesinos-Correa H, Bandala C, Carmona-Aparicio L, et al. Role of oxidative stress in refractory epilepsy: evidence in patients and experimental models. Int J Mol Sci. 2013 Jan 14;14(1):1455-76.
21. Forman HJ, Zhang H, Rinna A. Glutathione: overview of its protective roles, measurement, and biosynthesis. Mol Aspects Med. 2009 Feb;30(1-2):1-12.
22. Ahmed SS, Santosh W, Kumar S, Christlet HT. Metabolic profiling of Parkinson's disease: evidence of biomarker from gene expression analysis and rapid neural network detection. J Biomed Sci. 2009 Jul 13;16:63. doi: 10.1186/1423-0127-16-63.:63-16.
23. Dringen R, Gutterer JM, Hirrlinger J. Glutathione metabolism in brain metabolic interaction between astrocytes and neurons in the defense against reactive oxygen species. Eur J Biochem. 2000 Aug;267(16):4912-6.
24. Johnson WM, Wilson-Delfosse AL, Mieyal JJ. Dysregulation of glutathione homeostasis in neurodegenerative diseases. Nutrients. 2012 Oct 9;4(10):1399-440.
25. Ballatori N, Krance SM, Notenboom S, Shi S, Tieu K, Hammond CL. Glutathione dysregulation and the etiology and progression of human diseases. Biol Chem. 2009 Mar;390(3):191-214.
26. Kojovic M, Bologna M, Kassavetis P, Murase N, Palomar FJ, Berardelli A, et al. Functional reorganization of sensorimotor cortex in early Parkinson disease. Neurology. 2012 May 1;78(18):1441-8.
27. Limon-Pacheco JH, Hernandez NA, Fanjul-Moles ML, Gonsebatt ME. Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Radic Biol Med. 2007 Nov 1;43(9):1335-47.
28. Galvan A, Wichmann T. Pathophysiology of parkinsonism. Clin Neurophysiol. 2008 Jul;119(7):1459-74.
29. Robinson S, Basso G, Soldati N, Sailer U, Jovicich J, Bruzzone L, et al. A resting state network in the motor control circuit of the basal ganglia. BMC Neurosci. 2009 Nov 23;10:137. doi: 10.1186/1471-2202-10-137.:137-10.
30. Rommelfanger KS, Wichmann T. Extrastriatal dopaminergic circuits of the Basal Ganglia. Front Neuroanat. 2010;4:139. doi: 10.3389/fnana.2010.00139.:139.
31. Bronfeld M, Bar-Gad I. Loss of specificity in Basal Ganglia related movement disorders. Front Syst Neurosci. 2011 Jun 3;5:38. doi: 10.3389/fnsys.2011.00038. eCollection;%2011.:38.
32. Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441-66. doi: 10.1146/annurev-neuro-061010-113641.:441-66.
33. Obeso JA, Lanciego JL. Past, present, and future of the pathophysiological model of the Basal Ganglia. Front Neuroanat. 2011 Jul 12;5:39. doi: 10.3389/fnana.2011.00039. eCollection;%2011.:39.
34. Meredith GE, Kang UJ. Behavioral models of Parkinson's disease in rodents: a new look at an old problem. Mov Disord. 2006 Oct;21(10):1595-606.
35. Garcia-Munoz M, Carrillo-Reid L, Arbuthnott GW. Functional anatomy: dynamic States in Basal Ganglia circuits. Front Neuroanat. 2010 Nov 23;4:144. doi: 10.3389/fnana.2010.00144. eCollection;%2010.:144.
36. Alex KD, Pehek EA. Pharmacologic mechanisms of serotonergic regulation of dopamine neurotransmission. Pharmacol Ther. 2007 Feb;113(2):296-320.
37. Rice ME, Patel JC, Cragg SJ. Dopamine release in the basal ganglia. Neuroscience. 2011 Dec 15;198:112-37. doi: 10.1016/j.neuroscience.2011.08.066. Epub;%2011 Sep 14.:112-37.
38. Yao WD, Spealman RD, Zhang J. Dopaminergic signaling in dendritic spines. Biochem Pharmacol. 2008 Jun 1;75(11):2055-69.
39. Truong L, Allbutt H, Kassiou M, Henderson JM. Developing a preclinical model of Parkinson's disease: a study of behaviour in rats with graded 6-OHDA lesions. Behav Brain Res. 2006 Apr 25;169(1):1-9.
40. Vaarmann A, Gandhi S, Abramov AY. Dopamine induces Ca2+ signaling in astrocytes through reactive oxygen species generated by monoamine oxidase. J Biol Chem. 2010 Aug 6;285(32):25018-23.
41. Roberts RC, Roche JK, Conley RR, Lahti AC. Dopaminergic synapses in the caudate of subjects with schizophrenia: relationship to treatment response. Synapse. 2009 Jun;63(6):520-30.
42. Lee CR, Witkovsky P, Rice ME. Regulation of Substantia Nigra Pars Reticulata GABAergic Neuron Activity by H(2)O(2) via Flufenamic Acid-Sensitive Channels and K(ATP) Channels. Front Syst Neurosci. 2011 Abr 4;5:14. doi: 10.3389/fnsys.2011.00014. eCollection;%2011.:14.
43. Kreitzer AC, Malenka RC. Striatal plasticity and basal ganglia circuit function. Neuron. 2008 Nov 26;60(4):543-54.
44. Hutchison WD, Dostrovsky JO, Walters JR, Courtemanche R, Boraud T, Goldberg J, et al. Neuronal oscillations in the basal ganglia and movement disorders: evidence from whole animal and human recordings. J Neurosci. 2004 Oct 20;24(42):9240-3.
45. Wichmann T, Dostrovsky JO. Pathological basal ganglia activity in movement disorders. Neuroscience. 2011 Dec 15;198:232-44. doi: 10.1016/j.neuroscience.2011.06.048. Epub;% 2011 Jun 22.:232-44.
46. Mizuno Y, Hattori N, Kubo S, Sato S, Nishioka K, Hatano T, et al. Progress in the pathogenesis and genetics of Parkinson's disease. Philos Trans R Soc Lond B Biol Sci. 2008 Jun 27;363(1500):2215-27.
47. Gonzalez-Hernandez T, Cruz-Muros I, Afonso-Oramas D, Salas-Hernandez J, Castro-Hernandez J. Vulnerability of mesostriatal dopaminergic neurons in Parkinson's disease. Front Neuroanat. 2010 Oct 20;4:140. doi: 10.3389/fnana.2010.00140. eCollection; % 2010.:140.
48. Shukla V, Mishra SK, Pant HC. Oxidative stress in neurodegeneration. Adv Pharmacol Sci. 2011;2011:572634. doi: 10.1155/2011/572634. Epub;% 2011 Sep 21.:572634.
49. Nikam S, Nikam P, Ahaley SK, Sontakke AV. Oxidative stress in Parkinson's disease. Indian J Clin Biochem. 2009 Ene;24(1):98-101.
50. Hwang O. Role of oxidative stress in Parkinson's disease. Exp Neurobiol. 2013 Mar;22(1):11-7.
51. Garcia-Garcia A, Zavala-Flores L, Rodriguez-Rocha H, Franco R. Thiol-redox signaling, dopaminergic cell death, and Parkinson's disease. Antioxid Redox Signal. 2012 Dec 15;17(12):1764-84.
52. Kaur D, Lee D, Ragapolan S, Andersen JK. Glutathione depletion in immortalized midbrain-derived dopaminergic neurons results in increases in the labile iron pool: implications for Parkinson's disease. Free Radic Biol Med. 2009 Mar 1;46(5):593-8.
53. Hsu M, Srinivas B, Kumar J, Subramanian R, Andersen J. Glutathione depletion resulting in selective mitochondrial complex I inhibition in dopaminergic cells is via an NO-mediated pathway not involving peroxynitrite: implications for Parkinson's disease. J Neurochem 2005. Mar;92(5):1091-103.
54. Hoepken HH, Gispert S, Morales B, Wingerter O, Del TD, Mulsch A, et al. Mitochondrial dysfunction, peroxidation damage and changes in glutathione metabolism in PARK6. Neurobiol Dis. 2007 Feb;25(2):401-11.
55. Toffa S, Kunikowska GM, Zeng BY, Jenner P, Marsden CD. Glutathione depletion in rat brain does not cause nigrostriatal pathway degeneration. J Neural Transm. 1997;104(1):67-75.
56. Gao XF, Wang W, Yu Q, Burnstock G, Xiang ZH, He C. Astroglial P2X7 receptor current density increased following long-term exposure to rotenone. Purinergic Signal. 2011 Mar;7(1):65-72.
57. Garrido M, Tereshchenko Y, Zhevtsova Z, Taschenberger G, Bahr M, Kugler S, et al. Glutathione depletion and overproduction both initiate degeneration of nigral dopaminergic neurons. Acta Neuropathol. 2011 Apr;121(4):475-85.
58. Correa F, Ljunggren E, Mallard C, Nilsson M, Weber SG, Sandberg M, et al. The Nrf2-Inducible Antioxidant Defense in Astrocytes can be Both Up- and Down-Regulated by Activated Microglia: Involvement of p38 MAPK. Glia. 2011 May;59(5):785-99.
59. Wang X, Michaelis EK. Selective neuronal vulnerability to oxidative stress in the brain. Front Aging Neurosci. 2010;2:12. doi: 10.3389/fnagi.2010.00012.:12.
60. Higuchi Y. Glutathione depletion-induced chromosomal DNA fragmentation associated with apoptosis and necrosis. J Cell Mol Med. 2004 Oct;8(4):455-64.
61. Díaz-Hung ML, Blanco L, Pavon N, León R, Estupiñán B, Orta E, et al. Sensory-motor performance after acute glutathione depletion by L-buthionine sulfoximine injection into substantia nigra pars compacta. Behavioural Brain Research. 2014;271:286-93.
62. Avshalumov MV, Chen BT, Koos T, Tepper JM, Rice ME. Endogenous hydrogen peroxide regulates the excitability of midbrain dopamine neurons via ATP-sensitive potassium channels. J Neurosci. 2005 Apr 27;25(17):4222-31.
63. González ME, Fernández I, Bauza JY. Indicadores de estrés oxidativo en cerebros de ratas viejas con déficit cognitivo. Biotecnol Apl. 2007;24(2):145-50.
64. Smith MP, Cass WA. Oxidative stress and dopamine depletion in an intrastriatal 6-hydroxydopamine model of Parkinson's disease. Neuroscience. 2007 Feb 9;144(3):1057-66.
65. Circu ML, Yee AT. Glutathione and apoptosis. Free Radic Res. 2008 Aug;42(8):689-706.
66. Limon-Pacheco JH, Hernandez NA, Fanjul-Moles ML, Gonsebatt ME. Glutathione depletion activates mitogen-activated protein kinase (MAPK) pathways that display organ-specific responses and brain protection in mice. Free Radic Biol Med. 2007 Nov 1;43(9):1335-47.
67. De Bernardo S, Canals S, Casarejos MJ, Solano RM, Menendez J, Mena MA, et al. Role of extracellular signal-regulated protein kinase in neuronal cell death induced by glutathione depletion in neuron/glia mesencephalic cultures. J Neurochem. 2004 Nov;91(3):667-82.
Recibido: 5 de febrero de 2015.
Aprobado: 28 de febrero de 2015.
Mei-Li Díaz-Hung. Centro Internacional de Restauración Neurológica. Avenida 25 no. 15805, CP 11300. La Habana, Cuba.

