










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
Heberprot-P® es un medicamento obtenido a partir del Factor de crecimiento epidérmico humano recombinante (FCEhr) y es desarrollado por el Centro de Ingeniería Genética y Biotecnología (CIGB, 2006. La Habana, Cuba). Su administración está indicada en el tratamiento de las úlceras del pie en el diabético, para estimular la cicatrización y cierre de estas heridas. A diferencia de otras terapias establecidas para tratar la úlcera del pie diabético, este medicamento se indica en las úlceras más complejas que ya alcanzan los estadios 3 y 4 de la clasificación de Wagner.
Heberprot-P® es un medicamento liofilizado para inyección intralesional con el cual se superan las limitaciones de la aplicación tópica asociada a la presencia de actividad proteolítica en las heridas crónicas.1 Este modo de aplicación intralesional forma parte de una patente del CIGB aprobada en Cuba y otros 20 países como México, EE. UU., Sudáfrica, Europa, Japón, Brasil, Australia, entre otros.
El primer registro sanitario de Heberprot-P® se obtuvo en Cuba en el año 2006, lo cual permitió su comercialización en el territorio nacional. Su uso extendido en pacientes cubanos, así como la ejecución de estudios de poscomercialización, confirmaron los resultados de los estudios clínicos con este medicamento que fue seguro, con un 75 % de probabilidad de respuesta completa de granulación, el 61 % de cierre de la úlcera, el 16 % de reducción absoluta y el 71 % de reducción relativa del riesgo de amputación y la cantidad necesaria de pacientes a tratar para prevenir una amputación fue de seis. La relación riesgo beneficio es favorable al uso de Heberprot-P®.2,3,4,5,6,7,8 Estos resultados, con un importante aporte en la solución de un problema de salud a nivel mundial, condujeron al trabajo de registro sanitario en el exterior y actualmente Heberprot-P® está aprobado en otros 26 países, lo cual ha permitido el tratamiento de más de 392 037 pacientes donde se han reiterado los resultados vistos con los estudios en Cuba.
En los procesos de registro sanitario, de manera general, se conforma el expediente del producto según el formato del país y se recogen en él los elementos de calidad, seguridad y eficacia del producto a registrar,9 así como las informaciones legales correspondientes. El expediente se somete a la evaluación por la autoridad nacional reguladora. Durante el periodo de evaluación se solicitan completamientos de la documentación presentada, se hacen preguntas ante dudas de la autoridad evaluadora, se solicitan muestras del producto para evaluación analítica en los laboratorios del país, se solicita llevar a cabo la inspección de certificación de las buenas prácticas de fabricación y al final del proceso, se emite o no el certificado de Registro Sanitario en dependencia de las respuestas dadas y la valoración de la autoridad reguladora del país.
El proceso de registro sanitario en México para un producto biotecnológico, mostró importantes diferencias y particularidades respecto al antes descrito. Esto tiene por base la reforma legal que hubo en México a partir del año 2009, donde las normativas para el Registro Sanitario de biotecnológicos, sufrieron reformas y adiciones para fortalecer el marco regulatorio que los normaba.
Entre las adiciones antes referidas, la reglamentación mexicana incluyó el requisito de reunión con el Subcomité de Evaluación de Productos Biotecnológicos y el Comité de Moléculas Nuevas, para medicamentos biotecnológicos que se quieran comercializar en el territorio mexicano. Este paso define los medicamentos biotecnológicos que pueden presentar la solicitud de registro sanitario una vez que han pasado satisfactoriamente la evaluación ante ambos comités, conformados por especialistas y científicos en materia de biotecnología y que determinan si la información presentada, demuestra la seguridad, calidad y eficacia del medicamento.
Otra cualidad distintiva es que la reglamentación mexicana reconoce a los Terceros Autorizados, figura legal autorizada a evaluar un expediente de registro sanitario previo a la definitiva que hace la Autoridad Nacional Reguladora de México (Cofepris) y sobre cuya evaluación se generan ventajas tanto para la autoridad nacional reguladora, como para al solicitante del registro.
Estas y otras particularidades para el registro de productos biotecnológicos en México, nos hizo plantearnos una estrategia por pasos para lograr avanzar hasta la obtención del Registro Sanitario, cumpliendo cada requisito. De ahí que el objetivo de este trabajo sea describir el proceso de registro sanitario en México, del medicamento biotecnológico Heberprot-P® para el tratamiento de las úlceras del pie diabético.
Métodos
El proceso de registro sanitario de Heberprot-P® en México estuvo en conformidad con la Ley general de salud que reglamenta el derecho a la protección de la salud, establece las bases y modalidades para el acceso a los servicios de salud y la concurrencia de la Federación mexicana y las entidades federativas en materia de salubridad general.10) En conformidad, también, con el Reglamento de insumos para la salud que pauta el control sanitario de los insumos de uso y consumo humano y el de los establecimientos, actividades y servicios relacionados con estos insumos.11
Se revisó además la Farmacopea de los Estados Unidos Mexicanos,12) donde se consignan los métodos generales de análisis y los requisitos sobre identidad, pureza y calidad de los fármacos, aditivos, medicamentos y productos biológicos. Las Normas Oficiales Mexicanas se revisaron para temas específicos y peculiares de la reglamentación mexicana vigente13,14,15,16,17,18 por la necesidad de garantizar el cumplimiento de todo lo exigido para el registro sanitario en México.
La conformación del expediente de Registro Sanitario a presentarse ante Cofepris, primer paso en el proceso de registro sanitario, implicó la realización de acciones en función de obtener el documento exigido en dicho expediente o la información a incluir en él.
En la mayoría de estas acciones fue necesario presentar solicitudes ante Cofepris. Esta actividad la desempeñó Neuronic a través de los procedimientos establecidos. Cofepris publicaba el estado de cada solicitud en un sitio web habilitado para dar seguimiento a todo lo que recibía para evaluar.19) Una vez publicada la culminación de cada evaluación, Neuronic recibía las respuestas oficiales de la Cofepris. Todo el proceso de registro sanitario contó con la evaluación y seguimiento del responsable sanitario de Neuronic.
Las reuniones establecidas con el Subcomité de Evaluación de Productos Biotecnológicos y el Comité de Moléculas Nuevas, paso previo a la presentación de la solicitud de registro sanitario, se realizaron de conformidad con las guías, lineamientos y regulaciones, entre otros documentos, vigentes para esta actividad.20,21,22)
El CIGB se sometió a la visita de verificación por la parte mexicana para la certificación de las Buenas Prácticas de Fabricación (BPF) de acuerdo a su reglamentación.17,18,23
Se llevó a cabo la evaluación y conformación del expediente de Registro Sanitario por el procedimiento del Tercero Autorizado por la Secretaría de Salud mexicana, lo cual estuvo sujeto a lo dictaminado en la legislación mexicana.10,11,24,25 El titular del registro sanitario de Heberprot-P® en México contrató al Tercero Autorizado en Protección y Verificación Sanitaria (TAPVS) para esta actividad y la emisión del Informe Técnico Favorable. Una vez concluido el trabajo con el Tercero Autorizado, se presentó la solicitud de registro sanitario.
Resultados
El cuadro siguiente resume las acciones y resultados del proceso de registro sanitario de Heberprot-P® en México y a continuación, se detallan los aspectos esenciales de cada uno de los pasos establecidos.
Cuadro Proceso de registro sanitario de Heberprot-P® en México
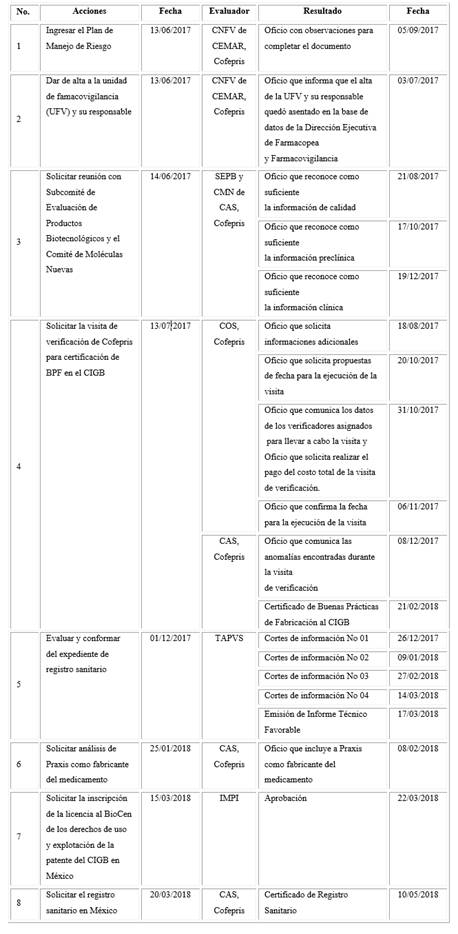
Cofepris: Autoridad Nacional Reguladora de México; CNFV: Centro Nacional de Farmacovigilancia; CEMAR: Comisión de Evidencia y Manejo de Riesgos; UFV: Unidad de Farmacovigilancia; SEPB: Subcomité de Evaluación de Productos Biotecnológicos; CMN: Comité de Moléculas Nuevas; COS: Comisión de Operación Sanitaria; CAS: Comisión de Autorización Sanitaria; BPF: Buenas Prácticas de Fabricación; TAPVS: Tercero Autorizado en Protección y Verificación Sanitaria; Praxis: fabricante registrado en España; BioCen: Centro Nacional de Biopreparados en Cuba; IMPI: Instituto Mexicano de la Propiedad Industrial.
Las primeras acciones fueron ingresar el Plan de Manejo de Riesgo y dar de alta a la unidad de farmacovigilancia (puntos 1 y 2 en el cuadro).
La reglamentación mexicana establece que el solicitante del registro sanitario ingrese al Centro Nacional de Farmacovigilancia, el Plan de Manejo de Riesgo para su revisión, previo a la solicitud de reunión técnica con los comités de evaluación. Estos comités emiten sus consideraciones y en caso de requerirse, hacen propuestas de modificación. Posterior a esto, el Centro Nacional entrega a Cofepris y al solicitante del registro sanitario, el dictamen final del Plan de Manejo de Riesgo, condición necesaria para la reunión con los comités evaluadores.
En junio de 2017 se ingresó ante la Comisión de Autorización Sanitaria de Cofepris la solicitud de reunión con dichos comités para la presentación de Heberprot-P® (punto 3 en el cuadro). Se incluyó además el expediente de registro sanitario del medicamento, previamente evaluado por el responsable sanitario de Neuronic.
Se realizaron tres reuniones ante diferentes grupos de expertos del Subcomité de Evaluación de Productos Biotecnológicos y el Comité de Moléculas Nuevas. En ellas se hicieron presentaciones del medicamento en los temas predefinidos de calidad, preclínica, clínica, farmacovigilancia, información para prescribir y patente y se dieron también, respuestas a las preguntas de los expertos. Con posterioridad a cada una, Cofepris emitió oficio con las conclusiones derivadas de la reunión donde consideró suficiente la información de calidad, de preclínica y de clínica respectivamente, realizó preguntas para completamiento a la información del medicamento y en el último oficio, de diciembre de 2017, se dieron las conclusiones derivadas de las reuniones sostenidas. Quedó cumplido el requisito de reunión y evaluación por ambos comités, por lo que fue posible continuar con los pasos establecidos, según las regulaciones mexicanas, para presentar la solicitud de Registro Sanitario de Heberprot-P®.
La próxima acción a realizar, fue la solicitud de la visita de verificación de Cofepris para la certificación de BPF en el CIGB (punto 4 en el cuadro). En febrero de 2018 se emitió el certificado de BPF a favor del CIGB para la producción de principios activos, acondicionamiento y empaque, control de calidad y liberación del medicamento.
Toda la información adicional a partir de las presentaciones ante los comités evaluadores y la certificación de BPF, se incluyó en el Registro Sanitario para su evaluación y conformación mediante el procedimiento del Tercero Autorizado por la Secretaría (punto 5 en el cuadro).
Se recibieron de TAPVS entre diciembre de 2017 y marzo de 2018, cuatro cortes de información con el estado de cumplimiento de los diferentes acápites. Se completaron por CIGB todas las informaciones solicitadas.
Temas específicos y peculiares de la reglamentación mexicana como la estabilidad, el material de envase y la farmacovigilancia, se debatieron más a fondo entre las partes involucradas y generó múltiples acciones por la parte cubana y la mexicana. La información exigida por Cofepris acerca de los aspectos mencionados, abarcó la colección de un volumen importante de evidencias para conocer la estabilidad del biofármaco, del medicamento y acerca del material de envase. La Unidad de Farmacovigilancia de Neuronic, se preparó para asumir y llevar a cabo las actividades de farmacovigilancia del Heberprot-P®. En marzo de 2018 TAVPS emitió el dictamen de Informe Técnico Favorable, preámbulo para poder presentar la solicitud de registro sanitario ante Cofepris.
Posterior al paso por los comités evaluadores, se decidió sustituir al fabricante del medicamento concebido inicialmente para el mercado mexicano, el Biocen. Se dispuso utilizar la planta de Praxis en España, en su lugar. Esta nueva situación requirió actualizar toda la información ante Cofepris para poder continuar el proceso de registro sanitario. En febrero de 2018 se emitió Oficio de Cofepris con la aprobación de la inclusión de Praxis como fabricante de Heberprot-P® (punto 6 en el cuadro).
En el punto 7 en el cuadro, se resume la acción donde el Biocen obtiene, mediante solicitud hecha por el CIGB al Instituto Mexicano de la Propiedad Industrial, la inscripción de la licencia para los derechos de uso y explotación de la patente del Heberprot-P® en México a partir de ser el titular del registro sanitario de Heberprot-P® en ese país.
Cofepris otorgó el Certificado de Registro Sanitario del Heberprot-P® en México el 10 mayo de 2018, donde reconoce al Heberprot-P® como medicamento biotecnológico innovador, en la categoría IV para su venta y suministro al público (punto 8 en el cuadro). Aquí se agrupan los medicamentos que para adquirirse requieren receta médica, pero que pueden proveerse (resurtirse, en México) tantas veces como lo indique el médico que prescriba. Así mismo se reconoció la estabilidad del medicamento en 24 meses y la vigencia del certificado por cinco años.
Discusión
El proceso de registro sanitario de Heberprot-P® en México transcurrió en once meses. Es importante notar que cada paso realizado fue requisito indispensable para poder continuar con el proceso. Los buenos resultados alcanzados por la parte cubana ante los comités evaluadores en los aspectos de calidad, preclínica, clínica, farmacovigilancia, información para prescribir, patente, así como en las respuestas ofrecidas y la pronta gestión realizada para subsanar debilidades y solucionar aspectos pendientes, fueron actividades clave que garantizaron la continuidad del proceso en dos aspectos esenciales para la obtención del registro sanitario en cuestión: la solicitud de visita de verificación de Cofepris para otorgar la certificación de BPF al CIGB y la participación de TAPVS, estilo de trabajo peculiar de México y que debe culminar con la emisión del Informe Técnico Favorable.
Previo a la emisión de ese Informe Técnico Favorable, el CIGB como fabricante del biofármaco (ingrediente farmacéutico activo o IFA), debió someterse a la visita de verificación para la certificación de BPF de acuerdo a la reglamentación mexicana. No así para Praxis (España) como fabricante del medicamento (Heberprot-P®).
El certificado de BPF del biofármaco y del medicamento debe ser emitido por la Autoridad mexicana o por la autoridad competente del país de origen. Si este último proviene de países con los cuales la Secretaría mexicana no tiene celebrados acuerdos de reconocimiento en materia de BPF, se lleva a cabo una visita de verificación para comprobar el cumplimiento de estas buenas prácticas. En el caso del medicamento fabricado en España, se dispone del certificado de BPF emitido por la autoridad de España que pertenece a la Agencia Europea de Medicamentos con la que existen los acuerdos antes señalados23) y Cofepris acepta dicha certificación como evidencia de cumplimiento de las BPF. En el caso del biofármaco fabricado en Cuba, se dispone del certificado de BPF emitido por la Autoridad Reguladora de Cuba (CECMED) con la que no existen, al día de hoy, los acuerdos antes mencionados.
La inspección se llevó cabo y las anomalías encontradas durante la visita de verificación se respondieron ante la Comisión de Autorización Sanitaria de Cofepris que la evaluó.
La reglamentación mexicana reconoce a los Terceros Autorizados, conformado legalmente por personas físicas o morales autorizadas por la Secretaría de Salud para emitir dictámenes respecto del cumplimiento de requisitos establecidos por dicha Secretaría o en las Normas Oficiales Mexicanas correspondientes, para efectos de trámites o autorizaciones sanitarias.10,11,24,25) En caso de que Cofepris reciba un Informe Técnico Favorable expedido por una institución reconocida como Tercero Autorizado, sus plazos de evaluación se reducen a la mitad.11).A esta ventaja, se suma el hecho de que las preguntas del Tercero Autorizado y el tiempo para responderlas no están sujeto a un número de veces o plazos establecidos, razones que condujeron a la decisión por la parte cubana de identificar y contratar a una de estas instituciones mexicanas para evaluar y conformar el expediente de Registro Sanitario de Heberprot-P®. En el procedimiento habitual de registro sanitario de medicamentos biotecnológicos innovadores, las solicitudes se responden en un plazo de 180 días naturales durante el cual, se puede solicitar información faltante, por única ocasión, durante los primeros 120 días naturales del plazo antes referido. El solicitante tiene un máximo de 100 días hábiles para responder.11
En el sentido práctico, este Informe Técnico Favorable, constituye una preevaluación del contenido del registro y contribuye a eliminar o minimizar posibles faltas u omisiones, antes de la entrega definitiva a la Autoridad Reguladora mexicana. El Tercero Autorizado emitió Informe Técnico Favorable (elaborado por ellos) tras un amplio proceso de revisión e intercambio con la parte cubana, además, el proyecto del certificado de Registro Sanitario.
La información exigida por Cofepris para aprobar a Praxis como fabricante del medicamento, demostraron que Praxis es un fabricante registrado al amparo de las leyes españolas, autorizado para la fabricación de medicamentos para uso humano y con cumplimiento de las BPF para productos biotecnológicos de conformidad con la licencia de fabricación y la certificación emitidas respectivamente, por la Agencia Española de Medicamentos y Productos Sanitarios. También se incluyeron evidencias de que Praxis es un fabricante autorizado en el Registro Sanitario de Heberprot-P® en Cuba emitido por la Autoridad Reguladora de Cuba (CECMED) y cuenta con experiencia en la fabricación del medicamento según los acuerdos de calidad entre CIGB y Praxis.
Se incluyeron, además, hechos que demostraron que la tecnología de fabricación se transfirió desde el CIGB a BioCen y finalmente a Praxis. Las evidencias entregadas demostraron que los procedimientos de operación, controles en proceso y especificaciones son equivalentes entre los tres sitios. Igualmente se incluyeron análisis comparativos entre Biocen-Praxis y CIGB-Praxis, así como las validaciones del proceso de fabricación en BioCen y Praxis que demostraron reproducibilidad en el proceso y cumplimiento con los criterios de aceptación. Praxis cuenta con estudios de estabilidad de conformidad con la Norma Oficial Mexicana NOM-073-SSA1-2015, evidencias que también se incluyeron.
Las especificaciones y los requisitos que recoge la reglamentación mexicana para los estudios de estabilidad de los biofármacos y medicamentos que se comercialicen en este territorio son parcialmente equivalente a los estándares internacionales,14 de manera que una parte de la información exigida, se encontraba en los protocolos e informes de estudio de estabilidad presentados a TAPVS.
Las evidencias enviadas para completar la otra parte de la información exigida, incluyeron los cromatogramas de la cromatografía líquida de alta resolución en fase reversa y las fotos de las electroforesis en gel de poliacrilamida con dodecilsulfato de sodio, de cada muestreo realizado a los lotes del biofármaco (ingrediente farmacéutico activo, Cuba) y de medicamentos (Heberprot-P®, Praxis, España), las órdenes de producción de los lotes sometidos a estudios de estabilidad, donde se verifican las materias primas empleadas, su número de lote y cantidad, los certificados de análisis del proveedor de las materias primas utilizadas y el fabricante correspondiente.
Para el material de envase, la norma mexicana no es equivalente a ninguna norma internacional15 por lo que los requisitos que establecen lo que debe contener el etiquetado de los medicamentos que se comercializan en el territorio mexicano y sus instructivos, se revisaron con profundidad. Para medicamentos biotecnológicos se exige la información para prescribir (IPP), en sus versiones amplia (IPPA) y reducida (IPPR),26) además de los marbetes para el instructivo y el etiquetado de las cajas y frasco ámpula. El Tercero Autorizado firma y acuña la propuesta de IPPA, IPPR y marbetes. La investigación exigida por México fue rigurosa. Los medicamentos biotecnológicos innovadores se identifican con las siglas MB y la información debe estar conforme a lo establecido en la Farmacopea de los Estados Unidos Mexicanos donde la nomenclatura de varios términos y conceptos es diferente a la de otros países donde se encuentra registrado el Heberprot-P®.
La farmacovigilancia en México tiene una estructura sólida conformada por ocho integrantes que hacen frente a las necesidades y demandas de salud pública en este territorio.10 Se profundizó en las acciones de dos de estos integrantes con impacto en el proceso de registro sanitario de Heberprot-P®: 1) El Centro Nacional de Farmacovigilancia, institución adscrita a Cofepris como parte de la Comisión de Evidencia y Manejo de Riesgos (CEMAR)27 y que se encarga de emitir las políticas y lineamientos para la operación de la farmacovigilancia en el territorio mexicano y da seguimiento y aprobación de varios documentos relacionados con esta actividad y 2) La Unidad de Farmacovigilancia (UFV), entidad dedicada a la implementación y desarrollo de actividades de farmacovigilancia de los medicamentos.
Otros temas como la titularidad del Registro Sanitario de Heberprot-P® en México, aspectos legales asociados a esto, la patente del producto en este país, así como su Denominación Distintiva, fueron medulares en el proceso de registro sanitario y llevaron análisis de mayor profundidad con la participación de todos los involucrados.
El Requisito de Planta en territorio mexicano para la fabricación y registro de medicamentos, permite tener al Titular del Registro Sanitario y a todos los fabricantes, en el extranjero y demanda tener en este caso a un representante legal con domicilio en México, como es Neuronic. El CIGB en Cuba cumplía con estas dos exigencias, pero hay una tercera que no puede cumplir.
Este tercer requisito implica que el registro sanitario y la patente del ingrediente activo tengan el mismo Titular o se cuente con la licencia correspondiente para el caso de la patente, ambas inscritas en el Instituto Mexicano de la Propiedad Industrial.11) Para ser Titular del Registro Sanitario de un medicamento en México se requiere además, en el caso de fabricantes extranjeros, contar con licencia, certificado o documento que acredite que la empresa cuenta con el permiso para fabricar medicamentos, expedido por la autoridad competente del país de origen.11). El CIGB, Titular del Registro Sanitario de Heberprot-P® en Cuba y varios países, está autorizado por CECMED para fabricar biofármacos (IFA; en el caso que nos ocupa) y llevar a cabo la operación de envase de los medicamentos que fabrica bajo contrato y, por tanto, no cumple con lo exigido en México para ser Titular de un Registro Sanitario.
El BioCen está autorizado a fabricar medicamentos en general, de ahí que para la titularidad se presentó a BioCen como fabricante de medicamentos para CIGB, con el aval exigido por Cofepris. Varios documentos contractuales se firmaron para garantizar la representación de BioCen por Neuronic ante las autoridades mexicanas; el suministro del biofármaco por CIGB a Praxis, bajo las condiciones de BioCen; la presencia y derechos del BioCen en la fabricación del medicamento en Praxis a partir del suministro del biofármaco por CIGB y en los elementos de calidad para esta actividad y la importación, distribución y comercialización del medicamento con el acuerdo de todas las partes involucradas.
En contraposición a esto, CIGB fue quien llevó a cabo la investigación y desarrollo de este producto, tiene todo el conocimiento sobre la molécula y es el que gerencia todos los procesos de registro sanitario de Heberprot-P® en el exterior. Esta problemática, se llevó ante Cofepris que escuchó positivamente todos los argumentos. La respuesta, sin embargo, estuvo apegada al cumplimiento de la reglamentación vigente en México donde CIGB no puede ser Titular del Registro Sanitario de Heberprot-P® en este país, a menos que tuviera un certificado emitido por CECMED avalando que es fabricante de medicamentos.
Heberprot-P® tuvo un primer Registro Sanitario en México que no se renovó y en el año 2017 se comenzó el proceso de registro sanitario que se describe en este trabajo. Existía con anterioridad el registro de la patente del producto en México a nombre del CIGB y, como se mencionó anteriormente, en la reglamentación mexicana vigente, el Titular del Registro Sanitario debe ser el Titular de la patente de la sustancia o ingrediente activo o debe contar con la licencia correspondiente, ambas inscritas en el Instituto Mexicano de la Propiedad Industrial.11) Fue necesario licenciar a BioCen los derechos de uso y explotación de la patente del CIGB en México y solicitar la inscripción del documento ante el Instituto Mexicano de la Propiedad Industrial, lo cual fue presentado y aprobado en marzo de 2018.
La Denominación Distintiva Heberprot-P®, marca comercial aprobada por Cofepris y con registro ante las autoridades competentes, ya existía en México dado que el primer registro sanitario se otorgó a una empresa con la que posteriormente hubo un proceso de terminación. Sobre este tema, la reglamentación mexicana plantea que no deberá usarse la misma Denominación Distintiva de otro medicamento con registro sanitario vigente, revocado o en trámite de registro, y que solo podrá utilizarse la misma Denominación Distintiva cuando se trate de diferentes formas farmacéuticas o diferentes dosis con un mismo principio activo y registrado por el mismo laboratorio.11). En el momento de iniciar este segundo proceso de registro sanitario, el primer registro ya no estaba vigente porque no se renovó. Para mantener la misma Denominación Distintiva, se presentó en el expediente de Registro Sanitario, la cesión de esta marca por el primer titular y una carta explicando que esta marca sería utilizada en México por el BioCen.
Se concluye que el proceso de registro sanitario de Heberprot-P® en México, a partir de las particularidades de la reglamentación mexicana para medicamentos biotecnológicos, incluyó trabajos con grupos de expertos antes de la solicitud oficial de Registro Sanitario a Cofepris. Todo esto permite a la autoridad mexicana hacer un trabajo más expedito basado en las evidencias de las evaluaciones realizadas que son parte de la información del registro sanitario. Como resultado de este proceso, se otorgó el registro sanitario a Heberprot-P® en mayo de 2018 y Cofepris lo reconoció como un medicamento biotecnológico innovador.

