










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.29 no.4 La Habana oct.-dic. 2012
REVIEW
Detrimental impact of acute and chronic glucose burden in wound-healing cells: fibroblasts, myofibroblasts and vascular precursor cells
Impacto perjudicial del exceso agudo y crónico de glucosa en células involucradas en la cicatrización: fibroblastos, miofibroblastos y células precursoras vasculares
Jorge Berlanga-Acosta1, Ernesto López-Mola1, Marianela Garcia-Siverio1, Gerardo Guillen-Nieto1, Pedro Lopez-Saura1, Calixto Valdez-Pérez2, Isabel Puentes-Madera2, William Savigne-Gutierrez2, Héctor Álvarez-Duarte2, Norberto Miranda-Espinosa3, Ana J Mir-Benítez4, Diana García del Barco1, Yssel Mendoza-Mari1, María D Martínez-Espina1, Ariana Garcia-Ojalvo1, Nelvys Subiros-Martinez1, Luis Herrera-Martínez1
1 Departamento de Cicatrización y Citoprotección. Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11 600, La Habana, Cuba.
2 Servicio de Angiopatía Diabética, Instituto Nacional de Angiología y Cirugía Vascular, INACV, Calzada del Cerro s/n. Cerro, La Habana, Cuba.
3 Departamento de Cirugía, Hospital Pediátrico Juan Manuel Márquez, Marianao, La Habana, Cuba.
4 Departamento de Cirugía Plástica y Reconstructiva, Hospital Joaquín Albarrán, Cerro, La Habana Cuba.
ABSTRACT
Type 2 diabetes mellitus comprises a group of non-communicable metabolic diseases with an expanding pandemic magnitude. Diabetes predisposes to lower extremities ulceration and impairs the healing process leading to wounds chronification. Diabetes also dismantles innate immunity favoring wound infection. Amputation is therefore acknowledged as one of the disease’s complications. Hyperglycemia appears as the proximal detonator of toxic effectors including pro-inflammation, spillover of reactive oxygen and nitrogen species. The systemic accumulation of advanced glycation end-products irreversibly impairs the entire physiology from cells-to-organs. Insulin axis deficiency weakens wounds’ anabolism and predisposes to inflammation. These factors converge to hamper fibroblasts and endothelial cells proliferation, migration, homing, secretion and organization of a productive granulation tissue. Diabetic wound bed may turn chronically inflamed, pro-catabolic and a superimposed source of circulating pro-inflammatory cytokines, establishing a self-perpetuating loop. Diabetic toxicity breadth includes mitochondrial damages in fibroblasts and endothelial cells becoming prone to apoptosis thus hindering granulation. Endothelial progenitor cells recruitment and tubulogenesis are also impaired. Failure of wound re-epithelialization remains as a clinical challenge while it appears to be biologically multifactorial. Novel medical interventions as the local intra-ulcer infiltration of epidermal growth factor have emerged to hopefully reduce the current worldwide amputation rates.
Keywords: diabetes, granulation, ulcer, re-epithelialization, growth factors.
RESUMEN
La diabetes mellitus tipo 2 implica desórdenes metabólicos no transmisibles, cuya incidencia va en aumento, con una extensión casi pandémica. Predispone a padecer úlceras en las extremidades inferiores y a su cronicidad, al afectar el proceso de cicatrización. También interfiere la inmunidad innata, lo que favorece la infección de posibles lesiones, y puede conducir a la amputación. La hiperglucemia desencadena los efectores tóxicos que incluyen la inflamación y la producción en exceso de especies reactivas de oxígeno y nitrógeno. Los productos finales altamente glicosilados, que se acumulan sistemáticamente, desarticulan la estructura de células y órganos. La deficiencia en el eje insulínico debilita el anabolismo en las lesiones y predispone a la inflamación. Estos factores convergen y debilitan la proliferación, la migración, el direccionamiento, la secreción y la organización de los fibroblastos y células endoteliales, lo que interfiere en la formación de tejido de granulación útil. El lecho de las heridas puede convertirse en una fuente inflamatoria y procatabólica de citocinas, y constituir un ciclo de perpetuación. La toxicidad diabética provoca daños mitocondriales en los fibroblastos y las células endoteliales, que los hace susceptibles de apoptosis y dificulta la granulación del tejido. También afecta el reclutamiento de las células progenitoras endoteliales, e impide la tubulogénesis. La regeneración del tejido epitelial en las lesiones sigue siendo un desafío clínico que depende de múltiples factores biológicos. Nuevas intervenciones médicas, como la infiltración local intralesional del factor de crecimiento epidérmico recombinante, prometen la reducción de las tasas mundiales de amputación.
Palabras clave: diabetes, granulación, úlcera, re-epitelización, factores de crecimiento.
INTRODUCTION
Diabetes mellitus represents today a worldwide pandemic disease under the most two common clinical forms identified as types 1 and 2. The former is a condition in which by autoimmune mechanisms pancreatic β-cells are eventually destroyed with an absolute insulin deficiency [1]. Type 2 diabetes mellitus (T2DM) is the most prevalent form of the disease and recently acknowledged not as a single clinical condition, but importantly, as a group of metabolic disorders. Diabetes in general, causes chronic hyperglycemia and a wide range of downstream metabolic disturbances and multi-organ complications [2]. It is notorious however, that although insulin secretion collapse, peripheral insulin resistance, and/or receptors’ activity failure play a definitive role for the onset of sustained hyperglycemia in T2DM, a large portion of body glucose is cleared by insulin-independent mechanisms, derived from the ability of plasma glucose to influence its own clearance by a mass action effect [3]. T2DM usually most common in adult subjects exhibits a slow, silent and insidious evolution. Hyperglycemia and its adjoined biochemical consequences undermine the whole tissues being sufficient to orchestrate irreversible systemic complications, from which the cells comprised in soft peripheral tissues and vascular structures do not escape. Lower extremities ulcerations and the potential for amputation are currently acknowledged as members of the list of diabetes complications [4].
Surgeon TD Pryce put forward as early as 1887 the link between diabetes and foot ulceration by writing in The Lancet that “[..] diabetes may of itself be a cause of perforating ulcer […]” [5]. However, despite the years of efforts and research, the pathogenesis of impaired wound healing in diabetes remains incompletely elucidated [6]. This poor-healing condition appears to be a multifactorial process which includes the amalgamation of systemic and local factors that ensure a perpetual forward loop up to chronification. Along this path, the cells seem to progressively wipe out their ability to trigger evolutionarily imprinted mechanisms as migration, proliferation and transdifferentiation, becoming increasingly static. Thus, diabetic wounds do not only become chronic by a concept of aberrant healing trajectory within a physiological time frame, but also by the asynchrony on the sequence of overlapping events that make up the tissue repair mega-process. Broadly speaking, diabetes impairs most if not all these events. Thus, the challenge that represents the diabetic wound healing failure is the clinical gross expression of an outstanding array of biochemical and cellular disorders [7]. These ideas are supported in the clinical arena by the alarming statistics of amputations around the world every year [8].
The healing process in diabetes is also jeopardized by the patient’s susceptibility to infection due to deficiencies on the innate immunity. Although the diabetic wound bed may be adversely overwhelmed by inflammatory cells, it does not represent an overt anti-bacterial protection. On the contrary, the diversion of glucose to the polyol pathway affects bacterial killing by reducing neutrophil opsonophagocytosis. Furthermore, hyperglycemia-induced reactive oxygen species (ROS) deregulate the innate immunity via an overactivation of the NF-κB transcription factor, thus amplifying the absurd inflammation and intoxicating the wound milieu [9, 10]. Peripheral arterial disease, leading to ischemia or lower limb hypoperfusion, is associated with the most severe outcomes, including lower probability of healing, longer healing times, higher probability of ulcer recurrence, greater risk of amputations, and potentially higher mortality [4]. Cells harvested and cultured from hypoperfused granulation tissues orchestrate a molecular program of arrest and senescence (Jorge Berlanga, manuscript in preparation). The outcome of the combination ‘healing failure’ and ‘infection susceptibility’ untowardly contributes to amputation.
Here we have reviewed the current evidences on the toxic resonance of acute and long term exposure to high glucose on the two main cells for the granulation tissue organization: fibroblasts and endothelial cells. We have included a characterization of the organizational disorders affecting diabetic granulation tissue and the challenge that represents its ultimate process, wound re-epithelialization. The literature search was based on English language articles downloaded from Pubmed [11] and Bioline International [12] databases.
CONSEQUENCES OF GLUCOSE OVERLOAD TOXICITY ON FIBROBLASTS AND ENDOTHELIAL CELLS
Fibroblasts
Fibroblasts are central to the wound healing process by secreting, contracting and remodeling the extracellular matrix (ECM). They also secrete growth factors as important messengers for mesenchymal-to-mesenchymal and epithelial-mesenchymal communication, especially for establishing the emerging basement membrane and subsequent re-epithelialization. Therefore, any impediment to fibroblast function is deterrent for normal wound healing and may result in chronic, non-healing wounds. The fibroblast, when engaged in fibrogenesis, displays the highly activated phenotype characteristic of myofibroblasts. Although their origin has not yet been definitely elucidated, proliferation of preexisting adjacent dermal fibroblasts, and probably recruited from the bone marrow, has been documented [13]. Under the high glucose burden imposed by diabetes, cutaneous and extracutaneous fibroblasts appear perturbed; and for many years, in vitro models recreating ‘clinical hyperglycemia’ have proved to disrupt normal fibroblasts physiology and derange the secretion of ECM ingredients. These experiments have suggested that high glucose concentration is the proximal detonator of a downstream cascade of molecular disturbances for the skin fibroblasts [14].
Rowe et al., who pioneered the in vitro models, demonstrated that the synthetic, proliferative and secreting capabilities are reduced in diabetics’ cutaneous fibroblasts [15]. Other parallel studies in which high glucose concentrations were introduced, proved to inhibit fibroblast proliferation, while the cells turned resistant to proliferate in response to growth factors such as insulin-like growth factor type-I (IGF-I) and epidermal growth factor (EGF) [16]. Following these attractive targets, Goldstein’s findings allowed for establishing the hypothesis that diabetics fibroblasts replicative life span did proportionally decline with diabetics predisposition under normal glucose concentrations, concluding that a persistent, heritable abnormality is present in mesenchymal tissues of overt diabetics and genetically predisposed subjects [17]. Years later, Goldstein also announced that cells obtained from insulin-dependent or insulin-independent diabetics not only exhibit abnormal replicative capacity in vitro, but that the aging process appeared more precociously than in non-diabetic counterparts [18]. Other studies showed that the addition of conditioned media from non-insulin-dependent diabetes mellitus wound fibroblasts induced a dose-dependent inhibition in normal fibroblast proliferation which appeared related to elevated L-lactate levels [19]. This replicative refractoriness of diabetic fibroblasts has been reproduced by different groups in subsequent years [20], thus confirming the need for additional external supplements to ensure cell cycle progression [21]. Accordingly, Loots et al. demonstrated the need of the simultaneous rather than sequential addition of different growth factors combinations for diabetic ulcer fibroblasts in order to induce a proliferative response [22]. Diabetic wound fibroblasts develop a quiescent and senescent phenotype, and their ability for horizontal and vertical migration is also dramatically impaired when compared to normal donor cells in different migration assay as in the modified Boyden chamber haptotaxis assay [23]. Most of these attributes are reproduced under acute exposures to high glucose concentrations so that migration speed is reduced by ~40%. This is associated to a decrease in cell directionality and to non-productive protrusive events (e.g.; loss of cell polarization), consistent with the increased activity of Rac1 and the projection of multiple lamellipodia. This experiment concluded that the generation of ROS may lie behind these abnormalities as they were partially or completely rescued by treatment with N-Acetyl-Cysteine (NAC) [2]. In contrast to the cellular reactions when exposed to high glucose in vitro, full-thickness wounds induced in non-diabetic pigs exposed to a local hyperglycemic environment exhibited no difference in wound closure when compared with normoglycemic controls [24]. This suggested that delayed wound healing by diabetes is a far more complex phenomenon than circumscribed to the high-glucose concentration itself [24]. As a consequence of the cutaneous accumulation of advanced glycation-end products (AGE), the skin increases its chronological age. One of the AGE precursors is 3-deoxyglucosone (3DG). Fibroblasts cultured on 3DG-treated collagen reduce the ability to migrate efficiently since 3DG increases its adherence to the matrix. Additionally, the authors described a higher level of misfolded proteins [25]. Using the same experimental system, this group demonstrated two years later that the inhibition in fibroblast migration, proliferation, and collagen expression by exposure to 3DG-collagen was mediated via extracellular regulated kinase 1/2 (ERK1/2) and the protein kinase B (Akt1) downregulation through activation of p38/MAPK (mitogen-activated protein kinase). These results indicated that p38 is a key signaling molecule that plays an opposite role during times of cellular growth and cellular stress [26]. Enriching the above findings, this group also demonstrated that 3DG-modified collagen induces oxidative stress, endoplasmic reticulum stress and apoptosis via caspase-3 activation. Oxidative stress appeared dependent on the upregulation of the NAD(P)H oxidase 4 (Nox4), a ROS Nox homologue, which appeared activated by p38/MAPK.
Proximal to this cascade is the effect caused by the interaction of the modified collagen with 3DG, which signals to the fibroblast by interacting with integrins alpha-1/beta-1 (α1β1) and not through the canonical AGE receptor (RAGE) [27]. Another group has also shown the induction of cutaneous fibroblasts apoptosis through cytoplasmic and mitochondrial pathways by plating the cells in an AGE-enriched environment made up by Nε-(carboxymethyl)lysine (CML)-collagen which primarily activated the classic RAGE [28]. A subsequent study elegantly demonstrated that after AGE-RAGE interaction, ROS generation increases, activating both nitrogen reactive species and ceramides, which in turn activates p38 and the c-Jun N-terminal protein kinase (c-JNK). Activated p38 and c-JNK triggers a cascade leading to amplified caspase-3 activity, whereas activation of Forkhead box O class 1 (Foxo1) increases the likelihood of apoptosis through enhanced expression of proapoptotic genes [29]. Under a number of circumstances, Foxo transcription factors induce the expression of BIM and other pro-apoptotic genes.
In addition to the deleterious effects of glucose and its derivatives, diabetic fibroblasts exhibit particular features. Literature documents that diabetic mice fibroblasts show a severe impairment in VEGF production under normoxic and hypoxic conditions as well as an increased pro-degradative activity due to the high expression of matrix metalloprotease type 9 (MMP-9) [30]. Similarly, diabetic pigs exhibited an impaired healing that was accompanied by a reduction of IGF-I in the wound milieu [24]. Studies with human fibroblasts have confirmed the pro-degradative phenotype by the increased MMP-2 and MMP-3 production and reduced collagens gene expression [31]. Human diabetic fibroblasts also showed a failure in nitric oxide (NO) production which was concomitant to elevations in MMP-8 and -9 [32]. The fact that these fibroblasts fail in secreting NO is particularly negative given its role for wound healing. Conversely, NO donors’ administration has shown to stimulate cell proliferation and restore the balance of MMPs [33].
It seems that amplification of oxidative stress acts as a primary culprit in harming fibroblasts biology in diabetes, involving electron transport in mitochondria. High intracellular glucose levels increase the electron transport chain in mitochondria during oxidative respiration, leading to formation of O2- and the generation of various ROS derivatives in the mitochondria. Other sources of oxidative stress in diabetes include glucose autoxidation, the polyol pathway with ensued depletion of anti-oxidant reserves and the formation of AGE [34]. Chronic hyperglycemia-induced mitochondrial ROS stimulate various signaling pathways that amplify inflammation and cell death. They include protein kinase C, c-JNK, and p38/MAPK [33]. According to an excellent review by Dana Graves’ group [35], ROS leads to the activation of members of the Foxo family. This is a family of transcription factors with apparently opposing roles that may defend cells against oxidative stress but also promote cell-cycle arrest in G1 by inducing the cell cycle inhibitory protein p27kip1 [36]. Foxo1 activation appears elevated in diabetic connective tissue cells and mediates AGE and tumor necrosis factor-alpha (TNF-α)-induced apoptosis, both of which are abundant in diabetic connective tissue [37]. Foxo1 limits wound healing by inhibiting fibroblasts proliferation and enhancing their apoptosis [37, 38]. Interestingly, insulin inactivates Foxo1 via Akt leading to its nuclear export and degradation. Defective insulin action in the skin has been proposed as an important mechanism contributing to wound healing defects in diabetes. Perhaps the assorted constellation of the hormone’s pharmacological bounties (increased expression of endothelial nitric oxide synthase (eNOS), vascular endothelial growth factor (VEGF) and stromal-derived factor-1α) observed in experimental and clinical wounds when insulin is topically administered, may be attributable to Foxo1 neutralization. Curiously, the acceleration of wound healing occurs in parallel to a local recovery in the expression of proteins involved in insulin signaling pathways [39]. Aside from the above arguments, these preclinical and clinical findings are not surprising in light of the potent anti-inflammatory, pro-anabolic and cytoprotective actions of insulin [40] which extend beyond the exclusive regulation of glucose homeostasis [3].
Despite the prolific investigation conducted so far, still questions remain to be answered in relation to ex vivo diabetics’ fibroblasts behavior:
- Why do diabetics’ fibroblasts evoke behavioral traits in culture, mirroring the donor’s tissue, even when grown under optimized oxygenation, nutrient, growth factors, and glucose supply?
- Is there any sort of ‘behavioral imprinting’ so that they are reminiscent from a diabetic donor?
- Why can cultured fibroblasts from both ischemic and neuropathic ulcers exhibit different ultrastructural morphology and organize the monolayer in a privative manner?
- Is there any epiphenomenon beyond the irreversible glycation sustaining the ‘impersonation’ of the in vivo traits?
Endothelial cells
Angiogenesis is a comprehensive term which indicates the physiological process involving the growth of new blood vessels or neovascularization. This is a vital process for embryological growth, tissue development, and wound healing. Different growth factors families regulate angiogenesis in collaboration with other proteins, by promoting endothelial cells recruitment, proliferation, migration, co-opting and collar stabilization. Among the growth factors are: VEGFs, fibroblast growth factor, angiopoietins, platelet-derived growth factor (PDGF) and transforming growth factor-β (TGF-β). Proteins comprise integrins, cadherins and ephrins. There is an enormous and ever-growing body of evidence indicating the close correlation between hyperglycemia and the abnormalities in endothelial function and morphology [41]. The UK Prospective Diabetes Study (UKPDS)[42] and Diabetes Control of Complications Trial (DCCT) [43] found microvascular disease and hyperglycaemia to be intrinsically related. Thus, anomalous angiogenesis is a hallmark of both type forms of diabetes which is clearly and early observable during the process of granulation tissue growth; condition that has been successfully reproduced in animal models [44]. For subjects with macrovascular disease, the defective angiogenesis prolongs and disturbs the healing process. The concept of abnormal angiogenesis extends beyond the wound, given the inability of these patients to create collateral circuits due to a VEGF-dependent monocytes dysfunction [45]. Furthermore, insulin has a dramatic impact on the endothelial homeostasis by its ability to stimulate NO release via a cascade that involves activation of the phosphatidylinositol 3-kinase (PI3K)-Akt signaling and eNOS phosphorylation. The later is of paramount importance in angiogenesis and wound healing as described below [46].
As depicted for fibroblasts, high glucose and the glycated by-products exert a dramatically toxic effect on endothelial cells and the vascular wall in general. In parallel, the endothelial cells per se seem to be a very sensitive target to high glucose. Endothelial dysfunction is intricately related to insulin resistance through the stimulatory effects of insulin on glucose disposal and NO production in the endothelium. Today, vascular dysfunction remains as a major cause of morbidity, amputation/disability and mortality in diabetic patients. Even after achieving the successful reperfusion of an ulcerated lower extremity, the healing process is slow and torpid. Therapeutic angiogenesis has been pursued for years but the clinical results have shown relatively limited outcomes [47-49].
High glucose concentrations have been associated with endothelial metabolic dysfunction in vitro and in vivo and as for multiple physiological processes; insulin and its downstream signaling regulate most of the endothelial cell functions [50]. High-glucose ambient has been shown to disturb endothelial cells cycle, increase DNA damage, delay endothelial cells replication and induce excessive cell death [51]. In addition, high glucose also prevents NO-induced inhibition of vascular smooth muscle cells migration [29] thus contributing to Monckeberg’s media thickening.
In vitro models simulating ‘normoglycemia’ and hyperglycemia have demonstrated that under high-glucose ambient, proliferation and tube formation of dermal microvascular endothelial cells appear impaired [52]. Furthermore, high glucose levels selectively trigger apoptosis in cultured endothelial cells as has been demonstrated by different laboratories [53]. High glucose induces the up-regulation of TNF-α level concomitant to the death receptors TNF-R1 and Fas in a variety of cultured endothelial cells [54]. Under this ambient, the expression of the proapoptotic Bax protein increases, cytochrome c is released, subsequently conjugating to the apoptotic protease activating factor-1 and triggering a caspase cascade-induced death [55].
Hyperglycemia-induced oxidative stress promotes inflammation through increased endothelial cells damage, microvascular permeability, and uncontrolled release of proinflammatory cytokines, including TNF-α, interlukin-1β (IL-1β), and interlukin-6 (IL-6), ultimately leading to decreased insulin sensitivity and diabetic vascular complications. Moreover, hyperglycemia-induced Foxo also plays an important role in the induction and amplification of proinflammatory cytokines production. Foxo1 directly binds to IL-1β promoter and increases its expression in macrophages [56].
Hyperglycemia and the accumulation of AGE disturb the role of angiogenic growth factors as VEGF, its receptor, its signaling pathway; thus disrupting endothelial proliferation, migration, and endothelial progenitor cells (EPCs) recruitment and release from bone marrow [57]. Insulin resistance disrupts the NO-mediated angiogenic positive regulation over angiogenic growth factors such as VEGF, fibroblast growth factor and TGF-β [57]. Studies using streptozotocin-induced diabetic mice with simultaneous hind-limb ischemia have suggested that the angiogenic responses remain preserved even under the diabetic state, and that 40 to 50% reduction of PDGF-BB expression is responsible for the induction of functional and morphological vascular abnormalities and pericytes apoptosis. Conversely, PDGF-BB external supplementation was sufficient to prevent limb autoamputation, also reproduced with a protein kinase C inhibitor that restored the expression of endogenous PDGF-BB [58].
The glycation of collagen and other proteins within the wound ECM and AGE accumulation bring catastrophic consequences for the angiogenic reaction with inhibition of angiogenesis in vivo. The fact that angiogenesis is restored by aminoguanidine treatment reinforces the antiangiogenic role of AGE [59]. Angiogenesis is a multifaceted process demanding an appropriate, non-glycated extracellular substrate. This is clearly illustrated by the fact that PDGF-BB anchors to different components of the ECM under physiological conditions acting as a natural depot and slow release system for the growth factor. Local PDGF unavailability has proved to impair the coverage of newly formed vessels with mural cells and local pericytes [60]. These evidences reinforce the pathophysiological impact of high glucose toxicity, the release of pro-inflammatory cytokines and the activation of the intrinsic mitochondrial-mediated apoptotic signaling pathway on endothelial cells. In summary, endothelial cells exposed to excess glucose develop a pro-inflammatory profile, becoming a significant source of cytokines and ROS production. The agonistic stimulation of the RAGE is able to mount the same response leading to apoptosis and vascular ruin. The pathogenic effects of hyperglycemia on fibroblasts and endothelial cells are summarized in figure 1.
Compelling evidences indicate that at least a portion of the hyperglycemia-mediated endothelial damages and dysfunctions are associated with an impaired mitochondrial activity resulting in mutations of mitochondrial DNA, due to a disproportionate ROS production, leading to an inflammatory reaction and apoptosis [61]. As a matter of fact, mitochondrial DNA has a much higher mutation rate than nuclear DNA because it lacks histones and is exposed to the direct action of oxygen radicals while its repair system is limited. Therefore, ROS appear to play a pivotal role in systemic endothelial deterioration and biological aging [62]. As described, ROS generation enhances Foxo1 activation and induction of several classes of genes that regulate endothelial cell behavior, including pro-inflammatory factors, and eventually the execution of apoptosis of endothelial and adjacent cells [35]. ROS-mediated lipid peroxidation appears to impair most healing events, contributing to growth factors reduction, keratinocytes migration failure, slow or torpid fibroplasia, delayed contraction and matrix remodeling, for not to mention abnormal angiogenesis [63]. Under experimental conditions, the pharmacological intervention with a chemical inhibitor of lipid peroxidation proved to reduce the local edema and to stimulate re-epithelialization, neovascularization, proliferation of fibroblasts, and synthesis and maturation of the ECM. A parallel finding was the normalization of VEGF mRNA expression and secretion in those diabetic mice. This further supports the view that lipid peroxidation perturbs VEGF production [64]. An extraordinary background has accumulated about the role of NO in vascular biology in diverse horizons as ischemia, inflammation and neovascularization. Impaired endothelium-dependent NO-mediated relaxation occurs in both cellular and in vivo models [65]. Many of the metabolic conditions associated with diabetes are conditioned by failure in NO synthesis or its degradation. In this respect, the integrity of the Akt/eNOS coupling pathway for a normal endothelial function appears compulsory [66]. Hyperglycemia is also associated to a deficit in tetrahydrobiopterin (BH4) and to an increase in arginase expression, which attempt against NO synthesis and normal endothelial functions such as vascular remodeling responses [67]. The increased generation of peroxynitrite levels under high glucose conditions contributed to deplete cellular anti-oxidant reserves as to activate the NF-κB transcription factor and consequently the expression of the inducible form of nitric oxide synthase (iNOS), intercellular adhesion molecule-1 and other inflammatory mediators [68].
EPCs are active players for the maintenance and repair of endothelial cells. They participate in angiogenesis as they proliferate, migrate and differentiate, and are a source for proangiogenic factors and cytokines [69]. Multiple evidences indicate that the number of circulating EPCs is decreased under both clinical forms of diabetes, which is likely to be involved in the pathogenesis of vascular complications [70]. Under experimental diabetic conditions the EPCs number appears significantly decreased in the bone marrow as in the peripheral blood which was reverted by treating the mice with insulin [71]. In general the bone marrow derived EPCs in the diabetic patients are considered as dysfunctional, producing fewer endothelial cells with reduced replicative, and migratory potential [72]. Tamarat et al. have described a limited capacity of diabetic animals-derived bone marrow mononuclear cell to differentiate into EPCs in vitro as to organize tubulogenic structures when subcutaneously implanted in a matrigel plug, thus hindering the revascularization of damaged areas [70]. Over again, the activation of p38/MAPK mediated by an excessive ROS generation has been identified as responsible for the EPCs impaired proangiogenic potential in vivo by limiting cell proliferation and differentiation [73]. To fully divert the physiological role of EPCs in tissue repair and angiogenesis, the duet hyperglycemia-ROS stimulates EPCs to produce pro-inflammatory cytokines and to shift NO production by elevating iNOS and decreasing eNOS [74]. As described for other cells, AGE treatment disrupts EPCs physiology thus leading to a downregulation of eNOS and the anti-apoptotic factor Bcl-2 expression, as well as an elevation in cyclooxygenase-2, proapoptotic factor Bax, NF-κB and caspase-3 in a MAPK (ERK/p38/c-JNK)-dependent manner [73].
Angiogenesis is of paramount importance for wound healing, but diabetes-mediated vascular cells damages are as varied and broad as it is the concept of systemic endothelial dysfunction. Diabetes distorts the angiogenic program so badly that angiogenic factors are deficitary where and when required (lower limbs), and overproduced in erroneous anatomical niches with fatal consequences for the patient (diabetic retinopathy).
FAILURE OF GRANULATION TISSUE ONSET AND PROGRESSION
Once described the main consequences of high glucose/hyperglycemia on the two principal architects of granulation tissue, fibroblasts and endothelial cells, the most distinguishing features for the onset of the granulation process in diabetic cutaneous wound healing will be recapitulated next.
Tissues’ regenerative capabilities have been neglected along the species evolution; thus, scarring process has emerged as an urgent alternative to favor the structural and functional restoration of a wounded zone. Within these events, the process of granulation tissue formation is pivotal as it constitutes a sort of living, temporary aggregate of cells and proteins, acting as a welding material until the tissue’s continuity is restored. However, the reluctance to trigger and sustain the out-growth of a productive granulation tissue with an appropriate ECM is typical in diabetic patients, and particularly if ischemia concurs. As mentioned, these wounds are characterized by a proliferative arrest, pro-inflamed, pro-oxidant and pro-degradative phenotype [75].
This stubbornness and slowness to heal in diabetes is conditioned by systemic and local factors that in complicity counteract intrinsic reparative mechanisms. In a broad systemic context, inflammation and anabolic deficit can be conceptually mentioned. Diabetic patients with foot ulceration bear a specific and ordered alteration of the immune status with an active upregulation of circulating levels of acute-phase proteins, cytokines, and chemokines that impose a chronic systemic inflammatory profile, and amplify local wound inflammatory networks [76]. The systemically elevated levels of pro-inflammatory response markers and the wound’s expression of cytokines and chemokines are among the culprits of the abnormal repair mechanism [77]. Another factor to be considered is that diabetes per se is a metabolic disease in which fuels metabolism is perturbed given the rupture of one of the most important anabolic axis of the organism: insulin/IGF-I. The role of insulin in wound healing is well known by its anabolic effect on wound protein balance, favoring synthesis and preventing degradation [78, 79]. IGF-I has a similar effect on stimulating wound tissue anabolism. Both insulin and IGF-I appear to act in part by the induction of the anabolic transcription factor ATF4 (CREB2), essential for the activation of the mammalian target of rapamycin complex 1 (m-TORC1) protein which in turn is required for protein synthesis via Foxo-dependent gene repression [80]. We do not rule out that the diabetes-concomitant deficit of incretins could participate in the negative anabolic balance observed in such wounds. Glucagon-like peptide-1 in addition to its anti-hyperglycemic actions is endowed with a vast number of multi-organ cytoprotective, trophic and anti-inflammatory effects [81]. In support to the glucagon-like peptide-1 action is the study by Ta et al. with alogliptin, a specific inhibitor of dipeptidyl peptidase-4, which was shown to inhibit macrophage-mediated inflammation response and was suggested as tissue remodeling promoter by inhibiting the expression of different MMPs [82].
Rapid formation and deposition of an appropriate ECM, in particular by fibroblasts, is required for an efficient cellular anchoring and homing at the wound bed. As mentioned above, the cutaneous fibroblast is a cell type sensitive to high glucose, AGE-precursors, AGE, ROS and TNF-α, rapidly undergoing premature senescence, arrest or apoptosis. Fibroblasts are the main source of collagen, and the number of fibroblasts can be taken as a measure of repair based on their collagen synthesis ability. It is very likely that the deficit of growth factors such as TGF-β1, IGF-I, and PDGF, that stimulate fibroblasts proliferation, transdifferentiation and the synthesis of matrix components, appear in shortage in diabetic foot ulcers (DFUs) and results in a scarce ECM formation. Numerous growth factors (TGF-β1, IGF-I, PDGF) are able to regulate the balanced expression of MMPs and tissue inhibitors of metalloproteases, while most of them exhibit an altered expression in DFUs [83]. Moreover, the imbalance in the DFU milieu between TGF-β1 and TGF-β3 in which the former appears downregulated, may explain fibroblasts quiescence in terms of proliferation and secretion [84]. This phenomenon represents the deficit of one of the most potent pro-fibrogenic and fibroblasts-mitogenic growth factors, which at the same time is able to downregulate macrophage activation [85].
The ECM represents the granulation tissue dynamic stroma that provides support for inflammatory cells, fibroblasts, and endothelial cells and allows for the chemotaxis of epithelial cells, thus hosting the re-epithelialization process [86]. One of the main challenges for the diabetic wound healing is the structuring of a normal matrix in quantity and quality. In general, a poor ECM formation distinguishes DFUs which can result from: i) diminished ECM synthesis, ii) increased ECM degradation rate by proteolytic enzymes, iii) toxicity due to glycated by-products accumulation, and iv) toxicity by biofilm bacterial contaminants diffusion [87]. We deem that an important cause of the clinical dilemma of the high rate of re-ulcerations and ipsilateral amputations in DFU patients’ shortly after re-epithelialization [88] may be inherent to the qualitative composition of the scar ECM to tolerate tensile forces and mechanical stress.
The diabetic granulation process does not generally exhibit the sequential cascade of events that characterize normal wound healing. This has been confirmed through the histopathological analysis of granulation tissue biopsies by Loots et al., who described the lesions as ‘frozen’ in a chronic low-grade inflammatory state associated to a scarce provisional ECM [89]. Our group’s serial biopsies from both neuropathic and ischemic ulcers-derived granulation tissue have identified histological differences for both types of wounds in the absence of clinical infection. Polymorphonuclear cells (PMN) infiltration is intense and prolonged particularly in neuropathic wounds, co-existing with a scarce ECM accumulation in which collagen deposit is impoverished (Figure 2).
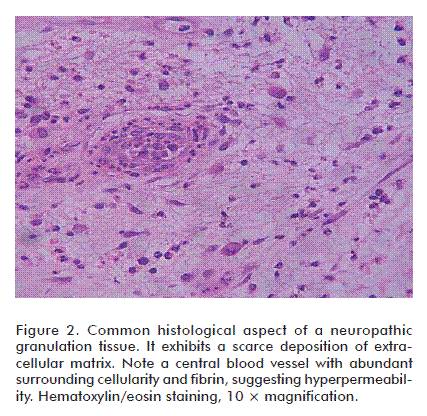
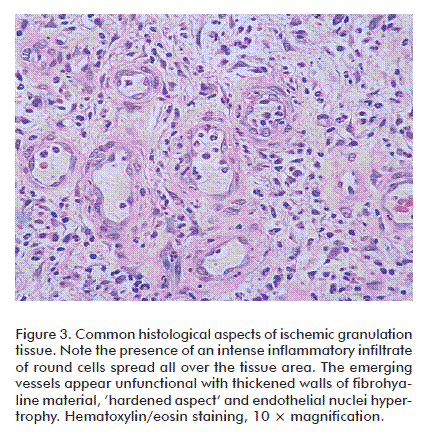
Under more mature stages, the neuropathics may also show an abnormal sprout of new small vessels and capillaries that may derive not from a normal angiogenic response but due to arterio-venous shunts. Our observations recall those of Black et al. who demonstrated that in neuropathic patients there exists a decrease in fibroblast proliferation and a scarce amount of collagen accumulation within the wound bed [90]. On the contrary, a broadly spread infiltration of round cells predominate in those patients suffering from wound bed ischemia, associated to a fibro-hyaline matrix of ‘hardened’ aspect and abnormal angiogenesis in which vascular wall cellular mosaicism, precocious media thickening, endothelial nuclei hypertrophy and many other defects can be identified (Figure 3). It is likely that the combination of arterial hypoperfusion and glucose toxic derivatives imprints a particular pattern of damage to the morphogenesis of vessels in the wound [91]. These observations incite to speculate that the biochemical microenvironment in ischemic and neuropathic diabetic wounds is different, and that the inflammatory ‘badge’ is in correspondence with the wound’s most prevalent pathogenic component [92]. In contrast to acute wounds in non-diabetic subjects, the inflammatory reaction in diabetics appears prolonged [93] and sharply delays granulation tissue formation and maturation [94]. Data derived from murine diabetic models indicate that the exaggerated inflammatory reaction is related to the prolonged expression of macrophage inflammatory protein-2 and macrophage chemoattractant protein-1 [95]. Furthermore, the downregulation of the anti-inflammatory cytokine IL-10 in DFUs environments represents the collapse of an important inflammatory restrainer [77]. Other evidences indicate that PMN are critical toward the acquisition and perpetuation of inflammation and a degradative phenotype. The granulocytes secrete TNF-α and IL-1β which act as a triggering signal for MMPs expression via the common NF-κB signaling pathway. Within the wound context, TNF-α stimulates its own secretion and that of IL-1β, contributing to a persistent inflammatory status [96]. TNF-α has proved to negatively impact the repair process as it is early secreted since the inflammatory phase. Its deregulation is not only associated with persistent inflammation but also to connective tissue degradation [97]. Concomitantly, TNF-α mediates its antagonistic effects on TGF-β1 through the c-JNK pathway via inhibition of Smad phosphorylation, consequently reducing the expression of TGF-β1, and that of several downstream matrix proteins [98]. In this highly proteolytic milieu, fibronectin, collagens, growth factors and their receptors are degraded while the wound is way down to a catabolic state [99]. Importantly, the perpetuated homing of PMN within the wound bed is associated to high local levels of elastase secretion, ROS and reactive nitrogen species [100]. High circulating and PMN-associated elastase levels are attributable to a poor glycemia control and are considered a risk marker for the development of diabetic angiopathy [101]. Fibronectin degradation, for instance, is referred as one among the several causes of diabetic re-epithelialization failure. Epidermal keratinocytes require of the interaction between fibronectin and its surface receptor integrin α5β1 to effectively migrate [102]. Curiously, insulin-degrading activity has also been demonstrated in the fluid of diabetic experimental and human wounds which have been shown to correlate with the glycated hemoglobin levels [103]. The connection between NO metabolism and foot ulcer proteases profile has been described. In contrast to elevated MMP-8 and -9 displayed by the non-healing diabetic foot wound, the concentration of NO appears significantly reduced. Diabetic skin fibroblasts treated with NO donor compounds selectively raised NO production, increased cell proliferation, and decreased the expression of MMP-8 and -9 in a dose-dependent manner. Thus, that NO resumes the cell proliferation program and promotes the reestablishment of an anti-proteases effect have emerged as argument in favor of the NO salutary effect in wound healing [32].
The link between wound cells and apoptosis was described above; we just wish to comment that in sharp contrast to wound-infiltrated inflammatory cells becoming refractory to apoptosis, granulation tissue-producing cells are sensitive to commit suicide where TNF-α stands as a major driving force. The negative impact of TNF-α levels on the sensitivity of tissues to insulin has been consistently documented. Skin cells are not excluded from this effect [104]. Conclusively, any therapeutic approach aimed to neutralize TNF-α or to increase the wound local availability of active TGF-β1 would be similarly effective for stimulating granulation tissue and wound closure [105].
Chronic wounds and especially diabetic foot ulcers exhibit a highly pro-oxidant microenvironment that amplifies the cytotoxic cascade. Endothelial cells and fibroblasts, in particular senescent fibroblasts, are a prominent source for oxygen radicals, but at the same time they turn into these radicals targets which, by converging mechanisms, arrest cell proliferation and induce apoptosis [106]. Thus, the disturbed oxidant/antioxidant balance as the AGE accumulation within the chronic wound microenvironment is considered a major factor amplifying the unrestrained and persistent inflammatory, toxic and catabolic state of non-healing wounds [100].
The failure of wound contraction is a clinical hallmark of diabetic granulation tissue. Fibroblast-to-myofibroblast transdifferentiation represents a key event during wound healing and tissue repair. The contractile force generated by myofibroblasts as a highly specialized cell, speeds the healing process of dermal wounds in healthy humans, accounting for an 80-90% of scar tissue reduction [107]. In addition, the contraction process reduces the area to be resurfaced by re-epithelialization which represents a sort of ergonomic response. In diabetic subjects however, contraction is impaired and deep ulcers heal by the combination of granulation and re-epithelialization. The classical view on dermal wound healing implies recruitment of local fibroblasts [108], followed by a subsequent process of transdifferentiation in which the fibroblasts gains a definitive phenotype of differentiated myofibroblasts by neo-expressing the α-smooth muscle actin (α-SMA). Nevertheless, α-SMA expression is precisely controlled by the joint action of growth factors like TGF-β1 and ECM proteins like the fibronectin splice variant ED-A, as by the local mechanical microenvironment [108]. It should be noted however, that indwelling fibroblasts in diabetic wounds are refractory to proliferate, adopt a senescent phenotype, and that TGF-β1, fibronectin and other matrix proteins may appear in deficit. Hence, all these factors may contribute to the poor contractile activity. Furthermore, Goldberg et al. showed that among the deleterious activities of TNF-α within the wound is to suppress α-SMA expression in human dermal fibroblasts [98]. Figure 4 integrates the cascade of deleterious factors that impact on diabetic granulation tissue onset.
If the animals-derived evidence that a high fraction of the wound myofibroblasts potentially derives from bone marrow fibrocytes is valid for humans [109]; then we have already learned that diabetes negatively impacts on the general bone marrow physiology [110], beyond this stromal-derived factor-1α (acting as recruiting factor) together with its CXCR4 chemokine receptor being also impaired [111]. Finally, it has been documented that the circulating acute inflammatory reactants involved in insulin resistance inhibit fibrocytes differentiation [112]. There are numerous cellular and molecular aspects unknown and that remain to be answered on the granulation tissue biology, such as: i) What are the molecular and cellular driving forces supporting the microscopic structural differences between neuropathic and ischemic ulcers beds?; ii) What is the explanation for the ‘inheritance’ of vascular changes as a dramatic Monckeberg media thickening in nascent arteries within an early hatching granulation tissue?; iii) Why granulation tissue is histomorphologically abnormal even in metabolically compensated patients?
Re-epithelialization at the clinical level is not a lesser important problem, as most of the diabetic wounds may granulate in time, while re-epithelialization is even far slower, arrhythmic, and torpid. Re-epithelialization is accomplished through the combined actions of keratinocytes’ dedifferentiation, proliferation and migration, requiring a complex basement membrane which emerges from the mutual interaction between mesenchymal and epithelial cells. Re-epithelialization failure is therefore one of the landmarks of diabetic and other chronic wounds. The epidermal edge of a chronic wound is thick and hyperproliferative, with mitotically active keratinocytes unable to migrate along the surface, and by the contrary, moving down deep into the neodermis. Therefore, it has been speculated that the non-healing edge keratinocytes do not successfully complete either the activation or the differentiation pathways. In consonance with this, one of the major issues in chronic wounds treatments is how to revert the chronic wound keratinocytes’ phenotype to a proper differentiating and migratory program [113].
Glucose has shown to exhibit a direct toxic effect on keratinocytes. As for other cells grown in the presence of high glucose concentrations, human epidermal keratinocytes significantly reduced their proliferation rate and replicative life span, and were rendered more susceptible to commit apoptosis [114]. Other studies also confirmed that hyperglycemic conditions abort keratinocytes’ proliferative ability and their migratory response [115]. Aside from the glucose-mediated direct cytotoxic effect on keratinocytes, AGE modification of type-I collagen and other ECM proteins impairs the integrin-mediated adhesion of keratinocytes to the basement matrix, and could thus contribute to the pathogenesis of diabetic re-epithelialization failure [116]. In this context, epithelial-mesenchymal interaction plays a role in establishing the profile and order of released factors regulating keratinocytes proliferation and differentiation [117].
The fact that insulin is biologically relevant for skin cells comes from the observation that insulin is an essential component for cultured human keratinocytes, demonstrating its involvement in the regulation of proliferation, survival, and metabolism [118]. Recent studies in this field document that, among other roles, insulin contributes to VEGF release in skin wound cells through an Akt1-mediated posttranscriptional mechanism [119]. Glucose is known to affect insulin action by regulating the expression of several genes including insulin receptor at both the transcriptional and translational levels [120]. Lack of insulin receptor expression derives in reduced skin proliferation and abnormal differentiation in vivo [121]. Furthermore, TNF-α has also been implicated in epithelial cells arrest by deeply perturbing critical elements of keratinocytes’ physiology, including insulin sensitivity [122].
A notorious study provided evidences on the roles of c-myc and β-catenin in impairing epithelial edges migration [123]. Nuclear β-catenin stabilization inhibits keratinocytes migration by blocking EGF response via c-myc induction, and repressing keratins 6 and 16 expression, ultimately depleting the pool of epidermal stem cells at the non-healing edge [123]. It is therefore evident that keratinocytes migration incapability plays an important role in re-epithelialization failure, since cytoskeletal keratins K2, K6 and K10 have been found diminished in DFUs [124]. Moreover, the observation that EGF response was blocked may have further deleterious impact. Many peptide growth factors, including members of the EGF family, accelerate wound re-epithelialization in vitro and in vivo [125]. Among them, the activation of the EGF family of ligands and its receptor (EGFR) is of physiological significance. Furthermore, EGFR expression is transiently increased at wound margins, suggesting its active role in wound repair. EGF stimulates both cell proliferation and motility [126], the later being dependent on EGFR autophosphorylation and the subsequent activation of phospholipase Cγ-1. On the other hand, EGFR activation also leads to membrane ruffling and focal adhesions through activation of members of the Rho subfamily of GTP-binding proteins [127]. Recent experiments document the negative effect of null mutation of the Slug transcription factor on its function as a downstream EGFR catalytic mediator for wound re-epithelialization. Thus, any interference with the EGFR cascade will hamper epithelial resurfacing [128]. Classic experiments provide illustrative examples on the relevance of the epithelial-mesenchymal cross-talk and on the irreplaceable role of growth factor as a networking bridge [129] for re-epithelialization. Skin-reconstitution studies have shown that bone marrow stromal cells, in addition to dermis-localized preadipocytes and fibroblasts, distinctively promote epidermal regeneration [130]. As diabetes courses with a deficient secretion of growth factors and other chemotactic mediators in areas of tissue repair, recruitment of circulating stromal cells appears reduced; which may turn into an additional hit to that of high glucose-associated toxicity [131].
At the end, there are so many factors which may interact as to obstruct chronic wounds re-epithelialization that it may turn into a puzzle. Above all, two questions from the clinical practice remain: Why do keratinocytes become stunted and arrested again soon after resuming migration following wound contours debridement? Why do the biopsies invariably show a hypertrophic lip of cells in vertical downward growth in spite of a horizontal polarization?
CONCLUDING REMARKS
Our current understanding on the molecular mechanisms impairing wound healing in diabetes simply sizes the tip of the iceberg. Diabetes per se is a complex disease. Even more complex is the group of hard-to-name members that constitutes the type 2 form. This is ethiopathogenically multifactorial and behaves as individual as the affected subject is. So, it is the pattern of clinical complications, including the wound itself. To date, all the evidences aim to high glucose burden as the proximal trigger to unleash acute and chronic self-perpetuating loops, which include but are not limited to ROS-lipid peroxidation, hyperinflammation/disimmunity, AGE-RAGE toxicity and mitochondrial dysfunction. All these factors enforce precocious senescence, arrest and apoptosis.
At both, experimental and clinical levels, the diabetic wound phenotype is the expression of countless molecular factors that orchestrate a complex biochemistry and an aberrant cellular behavior. The pathway to chronification has not been fully elucidated but by all means it represents a form of cells’ biological disobedience and entails the need of continuous surgical “cuttings” in order to transiently restore an acute behavior by ‘refreshing’ the cellular environment. Despite scientific and clinical efforts, and resources investments, amputation rates have not attained a significant reduction since the emergence of first line technology pharmaceuticals or sophisticated devices. Not to mention the dismal rates of local re-ulceration once the lesion was re-epithelialized. Since limb salvage is always a worthwhile goal and diabetic foot ulceration involves predisposing factors that can be diagnosed and clinically graded; primary care ulcer prevention plans, podiatric assistance, optimal glycemic control and educational programs, will be always far more socially rewarding.
REFERENCES
1. Daneman D. Type 1 diabetes. Lancet. 2006;367(9513):847-58.
2. Lamers ML, Almeida ME, Vicente-Manzanares M, Horwitz AF, Santos MF. High glucose-mediated oxidative stress impairs cell migration. PLoS One. 2011;6(8):e22865.
3. Bouche C, Serdy S, Kahn CR, Goldfine AB. The cellular fate of glucose and its relevance in type 2 diabetes. Endocr Rev. 2004;25(5):807-30.
4. Armstrong DG, Cohen K, Courric S, Bharara M, Marston W. Diabetic foot ulcers and vascular insufficiency: our population has changed, but our methods have not. J Diabetes Sci Technol. 2011;5(6):1591-5.
5. Pryce TD. A case of perforating ulcers of both feet associated with diabetes and ataxic symptoms. Lancet. 1887;2(3331):11-12.
6. Nguyen PD, Tutela JP, Thanik VD, Knobel D, Allen RJ, Jr., Chang CC, et al. Improved diabetic wound healing through topical silencing of p53 is associated with augmented vasculogenic mediators. Wound Repair Regen. 2010;18(6):553-9.
7. Berlanga-Acosta J, Valdéz-Pérez C, Savigne-Gutiérrez W, Mendoza-Marí Y, Franco-Pérez N, Vargas-Machiran E, et al. Cellular and molecular insights into the wound healing mechanism in diabetes. Biotecnol Apl. 2010;27(4):255-61.
8. Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet. 2005;366(9498):1719-24.
9. Gloire G, Legrand-Poels S, Piette J. NF-kappaB activation by reactive oxygen species: fifteen years later. Biochem Pharmacol. 2006;72(11):1493-505.
10. Mitra S, Abraham E. Participation of superoxide in neutrophil activation and cytokine production. Biochim Biophys Acta. 2006;1762(8):732-41.
11. Pubmed [Internet]. Bethesda, MD: National Center for Biotechnology Information, U.S. National Library of Medicine. c2012 [cited 2012 May 18]. Available from: www.ncbi.nlm.nih.gov/pubmed
12. Bioline International [Internet]. Sao Paulo: Reference Center on Environmental Information, CRIA, Brazil. c1989-2012 [cited 2012 May 18]. Available from: www.bioline.org.br
13. Guarino M, Tosoni A, Nebuloni M. Direct contribution of epithelium to organ fibrosis: epithelial-mesenchymal transition. Hum Pathol. 2009;40(10):1365-76.
14. Yevdokimova NY. High glucose-induced alterations of extracellular matrix of human skin fibroblasts are not dependent on TSP-1-TGFbeta1 pathway. J Diabetes Complications. 2003;17(6):355-64.
15. Rowe DW, Starman BJ, Fujimoto WY, Williams RH. Abnormalities in proliferation and protein synthesis in skin fibroblast cultures from patients with diabetes mellitus. Diabetes. 1977;26(4):284-90.
16. Hehenberger K, Hansson A. High glucose-induced growth factor resistance in human fibroblasts can be reversed by antioxidants and protein kinase C-inhibitors. Cell Biochem Funct. 1997;15(3):197-201.
17. Goldstein S, Moerman EJ, Soeldner JS, Gleason RE, Barnett DM. Diabetes mellitus and genetic prediabetes. Decreased replicative capacity of cultured skin fibroblasts. J Clin Invest. 1979;63(3):358-70.
18. Goldstein S. Cellular and molecular biological studies on diabetes mellitus. Pathol Biol (Paris). 1984;32(2):99-106.
19. Hehenberger K, Heilborn JD, Brismar K, Hansson A. Inhibited proliferation of fibroblasts derived from chronic diabetic wounds and normal dermal fibroblasts treated with high glucose is associated with increased formation of l-lactate. Wound Repair Regen. 1998;6(2):135-41.
20. Loots MA, Lamme EN, Mekkes JR, Bos JD, Middelkoop E. Cultured fibroblasts from chronic diabetic wounds on the lower extremity (non-insulin-dependent diabetes mellitus) show disturbed proliferation. Arch Dermatol Res. 1999;291(2-3):93-9.
21. Grazul-Bilska AT, Luthra G, Reynolds LP, Bilski JJ, Johnson ML, Adbullah SA, et al. Effects of basic fibroblast growth factor (FGF-2) on proliferation of human skin fibroblasts in type II diabetes mellitus. Exp Clin Endocrinol Diabetes. 2002;110(4):176-81.
22. Loots MA, Kenter SB, Au FL, van Galen WJ, Middelkoop E, Bos JD, et al. Fibroblasts derived from chronic diabetic ulcers differ in their response to stimulation with EGF, IGF-I, bFGF and PDGF-AB compared to controls. Eur J Cell Biol. 2002;81(3):153-60.
23. Xue SN, Lei J, Yang C, Lin DZ, Yan L. The biological behaviors of rat dermal fibroblasts can be inhibited by high levels of MMP9. Exp Diabetes Res. 2012;2012:494579.
24. Velander P, Theopold C, Hirsch T, Bleiziffer O, Zuhaili B, Fossum M, et al. Impaired wound healing in an acute diabetic pig model and the effects of local hyperglycemia. Wound Repair Regen. 2008;16(2):288-93.
25. Loughlin DT, Artlett CM. 3-Deoxyglucosone-collagen alters human dermal fibroblast migration and adhesion: implications for impaired wound healing in patients with diabetes. Wound Repair Regen. 2009;17(5):739-49.
26. Loughlin DT, Artlett CM. Modification of collagen by 3-deoxyglucosone alters wound healing through differential regulation of p38 MAP kinase. PLoS One. 2011;6(5):e18676.
27. Loughlin DT, Artlett CM. Precursor of advanced glycation end products mediates ER-stress-induced caspase-3 activation of human dermal fibroblasts through NAD(P)H oxidase 4. PLoS One. 2010;5(6):e11093.
28. Alikhani M, Maclellan CM, Raptis M, Vora S, Trackman PC, Graves DT. Advanced glycation end products induce apoptosis in fibroblasts through activation of ROS, MAP kinases, and the FOXO1 transcription factor. Am J Physiol Cell Physiol. 2007;292(2):C850-6.
29. Tong X, Ying J, Pimentel DR, Trucillo M, Adachi T, Cohen RA. High glucose oxidizes SERCA cysteine-674 and prevents inhibition by nitric oxide of smooth muscle cell migration. J Mol Cell Cardiol. 2008;44(2):361-9.
30. Lerman OZ, Galiano RD, Armour M, Levine JP, Gurtner GC. Cellular dysfunction in the diabetic fibroblast: impairment in migration, vascular endothelial growth factor production, and response to hypoxia. Am J Pathol. 2003;162(1):303-12.
31. Wall SJ, Sampson MJ, Levell N, Murphy G. Elevated matrix metalloproteinase-2 and -3 production from human diabetic dermal fibroblasts. Br J Dermatol. 2003;149(1):13-6.
32. Burrow JW, Koch JA, Chuang HH, Zhong W, Dean DD, Sylvia VL. Nitric oxide donors selectively reduce the expression of matrix metalloproteinases-8 and -9 by human diabetic skin fibroblasts. J Surg Res. 2007;140(1):90-8.
33. Sivitz WI, Yorek MA. Mitochondrial dysfunction in diabetes: from molecular mechanisms to functional significance and therapeutic opportunities. Antioxid Redox Signal. 2010;12(4):537-77.
34. Giacco F, Brownlee M. Oxidative stress and diabetic complications. Circ Res. 2010;107(9):1058-70.
35. Ponugoti B, Dong G, Graves DT. Role of forkhead transcription factors in diabetes-induced oxidative stress. Exp Diabetes Res. 2012;2012:939751.
36. Storz P. Forkhead homeobox type O transcription factors in the responses to oxidative stress. Antioxid Redox Signal. 2011;14(4):593-605.
37. Siqueira MF, Li J, Chehab L, Desta T, Chino T, Krothpali N, et al. Impaired wound healing in mouse models of diabetes is mediated by TNF-alpha dysregulation and associated with enhanced activation of forkhead box O1 (FOXO1). Diabetologia. 2010;53(2):378-88.
38. Obrenovich ME, Monnier VM. Apoptotic killing of fibroblasts by matrix-bound advanced glycation endproducts. Sci Aging Knowledge Environ. 2005;2005(4):pe3.
39. Lima MH, Caricilli AM, de Abreu LL, Araujo EP, Pelegrinelli FF, Thirone AC, et al. Topical insulin accelerates wound healing in diabetes by enhancing the AKT and ERK pathways: a double-blind placebo-controlled clinical trial. PLoS One. 2012;7(5):e36974.
40. Dandona P, Chaudhuri A, Ghanim H, Mohanty P. Insulin as an anti-inflammatory and antiatherogenic modulator. J Am Coll Cardiol. 2009;53(5 Suppl):S14-20.
41. Pirola L, Balcerczyk A, Okabe J, El-Osta A. Epigenetic phenomena linked to diabetic complications. Nat Rev Endocrinol. 2010;6(12):665-75.
42. Turner RC. The U.K. Prospective Diabetes Study. A review. Diabetes Care. 1998;21 Suppl 3:C35-8.
43. The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329(14):977-86.
44. Sonta T, Inoguchi T, Tsubouchi H, Sekiguchi N, Kobayashi K, Matsumoto S, et al. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37(1):115-23.
45. Waltenberger J, Lange J, Kranz A. Vascular endothelial growth factor-A-induced chemotaxis of monocytes is attenuated in patients with diabetes mellitus: A potential predictor for the individual capacity to develop collaterals. Circulation. 2000;102(2):185-90.
46. Efron DT, Most D, Barbul A. Role of nitric oxide in wound healing. Curr Opin Clin Nutr Metab Care. 2000;3(3):197-204.
47. Epstein SE, Kornowski R, Fuchs S, Dvorak HF. Angiogenesis therapy: amidst the hype, the neglected potential for serious side effects. Circulation. 2001;104(1):115-9.
48. Epstein SE, Fuchs S, Zhou YF, Baffour R, Kornowski R. Therapeutic interventions for enhancing collateral development by administration of growth factors: basic principles, early results and potential hazards. Cardiovasc Res. 2001;49(3):532-42.
49. Khan TA, Sellke FW, Laham RJ. Gene therapy progress and prospects: therapeutic angiogenesis for limb and myocardial ischemia. Gene Ther. 2003;10(4):285-91.
50. Duncan E, Ezzat V, Kearney M. Insulin and endothelial function: physiological environment defines effect on atherosclerotic risk. Curr Diabetes Rev. 2006;2(1):51-61.
51. Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88(2):E14-22.
52. Jain M, LoGerfo FW, Guthrie P, Pradhan L. Effect of hyperglycemia and neuropeptides on interleukin-8 expression and angiogenesis in dermal microvascular endothelial cells. J Vasc Surg. 2011;53(6):1654-60 e2.
53. Busik JV, Mohr S, Grant MB. Hyperglycemia-induced reactive oxygen species toxicity to endothelial cells is dependent on paracrine mediators. Diabetes. 2008;57(7):1952-65.
54. Rai NK, Suryabhan, Ansari M, Kumar M, Shukla VK, Tripathi K. Effect of glycaemic control on apoptosis in diabetic wounds. J Wound Care. 2005;14(6):277-81.
55. Kageyama S, Yokoo H, Tomita K, Kageyama-Yahara N, Uchimido R, Matsuda N, et al. High glucose-induced apoptosis in human coronary artery endothelial cells involves up-regulation of death receptors. Cardiovasc Diabetol. 2011;10:73.
56. Su D, Coudriet GM, Hyun Kim D, Lu Y, Perdomo G, Qu S, et al. FoxO1 links insulin resistance to proinflammatory cytokine IL-1beta production in macrophages. Diabetes. 2009;58(11):2624-33.
57. Simons M. Angiogenesis, arteriogenesis, and diabetes: paradigm reassessed? J Am Coll Cardiol. 2005;46(5):835-7.
58. Tanii M, Yonemitsu Y, Fujii T, Shikada Y, Kohno R, Onimaru M, et al. Diabetic microangiopathy in ischemic limb is a disease of disturbance of the platelet-derived growth factor-BB/protein kinase C axis but not of impaired expression of angiogenic factors. Circ Res. 2006;98(1):55-62.
59. Tamarat R, Silvestre JS, Huijberts M, Benessiano J, Ebrahimian TG, Duriez M, et al. Blockade of advanced glycation end-product formation restores ischemia-induced angiogenesis in diabetic mice. Proc Natl Acad Sci USA. 2003;100(14):8555-60.
60. Magnusson PU, Looman C, Ahgren A, Wu Y, Claesson-Welsh L, Heuchel RL. Platelet-derived growth factor receptor-beta constitutive activity promotes angiogenesis in vivo and in vitro. Arterioscler Thromb Vasc Biol. 2007;27(10):2142-9.
61. Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 2007;100(8):1128-41.
62. Fosslien E. Mitochondrial medicine-molecular pathology of defective oxidative phosphorylation. Ann Clin Lab Sci. 2001;31(1):25-67.
63. Martin A, Komada MR, Sane DC. Abnormal angiogenesis in diabetes mellitus. Med Res Rev. 2003;23(2):117-45.
64. Altavilla D, Saitta A, Cucinotta D, Galeano M, Deodato B, Colonna M, et al. Inhibition of lipid peroxidation restores impaired vascular endothelial growth factor expression and stimulates wound healing and angiogenesis in the genetically diabetic mouse. Diabetes. 2001;50(3):667-74.
65. Cooke JP, Losordo DW. Nitric oxide and angiogenesis. Circulation. 2002;105(18):2133-5.
66. Leo CH, Hart JL, Woodman OL. Impairment of both nitric oxide-mediated and EDHF-type relaxation in small mesenteric arteries from rats with streptozotocin-induced diabetes. Br J Pharmacol. 2011;162(2):365-77.
67. Kampfer H, Pfeilschifter J, Frank S. Expression and activity of arginase isoenzymes during normal and diabetes-impaired skin repair. J Invest Dermatol. 2003;121(6):1544-51.
68. Cooke CL, Davidge ST. Peroxynitrite increases iNOS through NF-kappaB and decreases prostacyclin synthase in endothelial cells. Am J Physiol Cell Physiol. 2002;282(2):C395-402.
69. Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J. 2009;73(3):411-8.
70. Tamarat R, Silvestre JS, Le Ricousse-Roussanne S, Barateau V, Lecomte-Raclet L, Clergue M, et al. Impairment in ischemia-induced neovascularization in diabetes: bone marrow mononuclear cell dysfunction and therapeutic potential of placenta growth factor treatment. Am J Pathol. 2004;164(2):457-66.
71. Saito H, Yamamoto Y, Yamamoto H. Diabetes alters subsets of endothelial progenitor cells that reside in blood, bone marrow, and spleen. Am J Physiol Cell Physiol. 2012;302(6):C892-901.
72. Loomans CJ, de Koning EJ, Staal FJ, Rookmaaker MB, Verseyden C, de Boer HC, et al. Endothelial progenitor cell dysfunction: a novel concept in the pathogenesis of vascular complications of type 1 diabetes. Diabetes. 2004;53(1):195-9.
73. Shen C, Li Q, Zhang YC, Ma G, Feng Y, Zhu Q, et al. Advanced glycation endproducts increase EPC apoptosis and decrease nitric oxide release via MAPK pathways. Biomed Pharmacother. 2010;64(1):35-43.
74. Reinhard H, Jacobsen PK, Lajer M, Pedersen N, Billestrup N, Mandrup-Poulsen T, et al. Multifactorial treatment increases endothelial progenitor cells in patients with type 2 diabetes. Diabetologia. 2010;53(10):2129-33.
75. Acosta JB, del Barco DG, Vera DC, Savigne W, Lopez-Saura P, Guillen Nieto G, et al. The pro-inflammatory environment in recalcitrant diabetic foot wounds. Int Wound J. 2008;5(4):530-9.
76. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. 2011;186(2):1162-72.
77. Weigelt C, Rose B, Poschen U, Ziegler D, Friese G, Kempf K, et al. Immune mediators in patients with acute diabetic foot syndrome. Diabetes Care. 2009;32(8):1491-6.
78. Tuvdendorj D, Zhang XJ, Chinkes DL, Aarsland A, Kulp GA, Jeschke MG, et al. Intensive insulin treatment increases donor site wound protein synthesis in burn patients. Surgery. 2011;149(4):512-8.
79. Zhang XJ, Chinkes DL, Irtun O, Wolfe RR. Anabolic action of insulin on skin wound protein is augmented by exogenous amino acids. Am J Physiol Endocrinol Metab. 2002;282(6):E1308-15.
80. Adams CM. Role of the transcription factor ATF4 in the anabolic actions of insulin and the anti-anabolic actions of glucocorticoids. J Biol Chem. 2007;282(23):16744-53.
81. Gupta V. Pleiotropic effects of incretins. Indian J Endocrinol Metab. 2012;16 Suppl 1:S47-56.
82. Ta NN, Li Y, Schuyler CA, Lopes-Virella MF, Huang Y. DPP-4 (CD26) inhibitor alogliptin inhibits TLR4-mediated ERK activation and ERK-dependent MMP-1 expression by U937 histiocytes. Atherosclerosis. 2010;213(2):429-35.
83. Galkowska H, Wojewodzka U, Olszewski WL. Chemokines, cytokines, and growth factors in keratinocytes and dermal endothelial cells in the margin of chronic diabetic foot ulcers. Wound Repair Regen. 2006;14(5):558-65.
84. Blakytny R, Jude EB. Altered molecular mechanisms of diabetic foot ulcers. Int J Low Extrem Wounds. 2009;8(2):95-104.
85. Surh CD, Sprent J. TGF-beta puts the brakes on homeostatic proliferation. Nat Immunol. 2012;13(7):628-30.
86. Daley WP, Peters SB, Larsen M. Extracellular matrix dynamics in development and regenerative medicine. J Cell Sci. 2008;121(Pt 3):255-64.
87. Schultz G, Berlanga-Acosta J, Cowan L, Stechmiller J. Linking the Advanced Glycation Endproducts/Receptor for Advanced Glycation Endproducts Pathway in Diabetics with Inflammation and Topical Antiinflammatory Treatments of Chronic Wounds. In: Sen CK, editor. Advances in Wound Care. Volume 1. Ohio: The Ohio State University Medical Center; 2010. p. 248-52.
88. Skoutas D, Papanas N, Georgiadis GS, Zervas V, Manes C, Maltezos E, et al. Risk factors for ipsilateral reamputation in patients with diabetic foot lesions. Int J Low Extrem Wounds. 2009;8(2):69-74.
89. Loots MA, Lamme EN, Zeegelaar J, Mekkes JR, Bos JD, Middelkoop E. Differences in cellular infiltrate and extracellular matrix of chronic diabetic and venous ulcers versus acute wounds. J Invest Dermatol.1998;111(5):850-7.
90. Black E, Vibe-Petersen J, Jorgensen LN, Madsen SM, Agren MS, Holstein PE, et al. Decrease of collagen deposition in wound repair in type 1 diabetes independent of glycemic control. Arch Surg. 2003;138(1):34-40.
91. Sliman SM, Eubank TD, Kotha SR, Kuppusamy ML, Sherwani SI, Butler ES, et al. Hyperglycemic oxoaldehyde, glyoxal, causes barrier dysfunction, cytoskeletal alterations, and inhibition of angiogenesis in vascular endothelial cells: aminoguanidine protection. Mol Cell Biochem. 2010;333(1-2):9-26.
92. Berlanga-Acosta J, Schultz GS, Lopez-Saura P. Biology of the Diabetic Wound. In: Overhaussen PE, editor. Foot Ulcers: Causes, Diagnosis and Treatments. Hauppauge: Nova Science Publishers, Inc.; 2009. p. 51-88.
93. Mulder GD, Lee DK, Jeppesen NS. Comprehensive review of the clinical application of autologous mesenchymal stem cells in the treatment of chronic wounds and diabetic bone healing. Int Wound J. 2012. Forthcoming.
94. Usui ML, Mansbridge JN, Carter WG, Fujita M, Olerud JE. Keratinocyte migration, proliferation, and differentiation in chronic ulcers from patients with diabetes and normal wounds. J Histochem Cytochem. 2008;56(7):687-96.
95. Wetzler C, Kampfer H, Stallmeyer B, Pfeilschifter J, Frank S. Large and sustained induction of chemokines during impaired wound healing in the genetically diabetic mouse: prolonged persistence of neutrophils and macrophages during the late phase of repair. J Invest Dermatol. 2000;115(2):245-53.
96. Nwomeh BC, Yager DR, Cohen IK. Physiology of the chronic wound. Clin Plast Surg. 1998;25(3):341-56.
97. Naguib G, Al-Mashat H, Desta T, Graves DT. Diabetes prolongs the inflammatory response to a bacterial stimulus through cytokine dysregulation. J Invest Dermatol. 2004;123(1):87-92.
98. Goldberg MT, Han YP, Yan C, Shaw MC, Garner WL. TNF-alpha suppresses alpha-smooth muscle actin expression in human dermal fibroblasts: an implication for abnormal wound healing. J Invest Dermatol. 2007;127(11):2645-55.
99. Mast BA, Schultz GS. Interactions of cytokines, growth factors, and proteases in acute and chronic wounds. Wound Repair Regen. 1996;4(4):411-20.
100. Weiss SJ. Tissue destruction by neutrophils. N Engl J Med. 1989;320(6):365-76.
101. Piwowar A, Knapik-Kordecka M, Warwas M. Concentration of leukocyte elastase in plasma and polymorphonuclear neutrophil extracts in type 2 diabetes. Clin Chem Lab Med. 2000;38(12):1257-61.
102. Stanley CM, Wang Y, Pal S, Klebe RJ, Harkless LB, Xu X, et al. Fibronectin fragmentation is a feature of periodontal disease sites and diabetic foot and leg wounds and modifies cell behavior. J Periodontol. 2008;79(5):861-75.
103. Duckworth WC, Fawcett J, Reddy S, Page JC. Insulin-degrading activity in wound fluid. J Clin Endocrinol Metab. 2004;89(2):847-51.
104. Mantzoros CS, Moschos S, Avramopoulos I, Kaklamani V, Liolios A, Doulgerakis DE, et al. Leptin concentrations in relation to body mass index and the tumor necrosis factor-alpha system in humans. J Clin Endocrinol Metab. 1997;82(10):3408-13.
105. Mi Q, Riviere B, Clermont G, Steed DL, Vodovotz Y. Agent-based model of inflammation and wound healing: insights into diabetic foot ulcer pathology and the role of transforming growth factor-beta1. Wound Repair Regen. 2007;15(5):671-82.
106. Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51(1):198-203.
107. Petrova N, Edmonds M. Emerging drugs for diabetic foot ulcers. Expert Opin Emerg Drugs. 2006;11(4):709-24.
108. Desmouliere A, Chaponnier C, Gabbiani G. Tissue repair, contraction, and the myofibroblast. Wound Repair Regen. 2005;13(1):7-12.
109. Mori L, Bellini A, Stacey MA, Schmidt M, Mattoli S. Fibrocytes contribute to the myofibroblast population in wounded skin and originate from the bone marrow. Exp Cell Res. 2005;304(1):81-90.
110. Fadini GP, Avogaro A. It is all in the blood: the multifaceted contribution of circulating progenitor cells in diabetic complications. Exp Diabetes Res. 2012;2012:742976.
111. Bermudez DM, Xu J, Herdrich BJ, Radu A, Mitchell ME, Liechty KW. Inhibition of stromal cell-derived factor-1alpha further impairs diabetic wound healing. J Vasc Surg. 2011;53(3):774-84.
112. Pilling D, Buckley CD, Salmon M, Gomer RH. Inhibition of fibrocyte differentiation by serum amyloid P. J Immunol. 2003;171(10):5537-46.
113. Morasso MI, Tomic-Canic M. Epidermal stem cells: the cradle of epidermal determination, differentiation and wound healing. Biol Cell. 2005;97(3):173-83.
114. Deveci M, Gilmont RR, Dunham WR, Mudge BP, Smith DJ, Marcelo CL. Glutathione enhances fibroblast collagen contraction and protects keratinocytes from apoptosis in hyperglycaemic culture. Br J Dermatol. 2005;152(2):217-24.
115. Lan CC, Liu IH, Fang AH, Wen CH, Wu CS. Hyperglycaemic conditions decrease cultured keratinocyte mobility: implications for impaired wound healing in patients with diabetes. Br J Dermatol. 2008;159(5):1103-15.
116. Zhu P, Yang C, Chen LH, Ren M, Lao GJ, Yan L. Impairment of human keratinocyte mobility and proliferation by advanced glycation end products-modified BSA. Arch Dermatol Res. 2011;303(5):339-50.
117. El Ghalbzouri A, Hensbergen P, Gibbs S, Kempenaar J, van der Schors R, Ponec M. Fibroblasts facilitate re-epithelialization in wounded human skin equivalents. Lab Invest. 2004;84(1):102-12.
118. Liu Y, Petreaca M, Yao M, Martins-Green M. Cell and molecular mechanisms of keratinocyte function stimulated by insulin during wound healing. BMC Cell Biol. 2009;10:1.
119. Goren I, Muller E, Schiefelbein D, Gutwein P, Seitz O, Pfeilschifter J, et al. Akt1 controls insulin-driven VEGF biosynthesis from keratinocytes: implications for normal and diabetes-impaired skin repair in mice. J Invest Dermatol. 2009;129(3):752-64.
120. Terashi H, Izumi K, Deveci M, Rhodes LM, Marcelo CL. High glucose inhibits human epidermal keratinocyte proliferation for cellular studies on diabetes mellitus. Int Wound J. 2005;2(4):298-304.
121. Wertheimer E, Spravchikov N, Trebicz M, Gartsbein M, Accili D, Avinoah I, et al. The regulation of skin proliferation and differentiation in the IR null mouse: implications for skin complications of diabetes. Endocrinology. 2001;142(3):1234-41.
122. Banno T, Gazel A, Blumenberg M. Effects of tumor necrosis factor-alpha (TNF alpha) in epidermal keratinocytes revealed using global transcriptional profiling. J Biol Chem. 2004;279(31):32633-42.
123. Stojadinovic O, Brem H, Vouthounis C, Lee B, Fallon J, Stallcup M, et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. 2005;167(1):59-69.
124. Sivamani RK, Garcia MS, Isseroff RR. Wound re-epithelialization: modulating keratinocyte migration in wound healing. Front Biosci. 2007;12:2849-68.
125. Coulombe PA. Wound epithelialization: accelerating the pace of discovery. J Invest Dermatol. 2003;121(2):219-30.
126. Nanney LB, Paulsen S, Davidson MK, Cardwell NL, Whitsitt JS, Davidson JM. Boosting epidermal growth factor receptor expression by gene gun transfection stimulates epidermal growth in vivo. Wound Repair Regen. 2000; 8(2):117-27.
127. Li S, Wang Q, Wang Y, Chen X, Wang Z. PLC-gamma1 and Rac1 coregulate EGF-induced cytoskeleton remodeling and cell migration. Mol Endocrinol. 2009;23(6):901-13.
128. Kusewitt DF, Choi C, Newkirk KM, Leroy P, Li Y, Chavez MG, et al. Slug/Snai2 is a downstream mediator of epidermal growth factor receptor-stimulated reepithelialization. J Invest Dermatol. 2009;129(2):491-5.
129. Frank S, Hubner G, Breier G, Longaker MT, Greenhalgh DG, Werner S. Regulation of vascular endothelial growth factor expression in cultured keratinocytes. Implications for normal and impaired wound healing. J Biol Chem. 1995;270(21):12607-13.
130. Aoki S, Toda S, Ando T, Sugihara H. Bone marrow stromal cells, preadipocytes, and dermal fibroblasts promote epidermal regeneration in their distinctive fashions. Mol Biol Cell. 2004;15(10):4647-57.
131. Gopalakrishnan V, Vignesh RC, Arunakaran J, Aruldhas MM, Srinivasan N. Effects of glucose and its modulation by insulin and estradiol on BMSC differentiation into osteoblastic lineages. Biochem Cell Biol. 2006; 84(1):93-101.
Received in October, 2012.
Accepted in November, 2012.
Jorge Berlanga-Acosta. Departamento de Cicatrización y Citoprotección. Dirección de Investigaciones Biomédicas, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/ 158 y 190, Cubanacán, Playa, CP 11 600, La Habana, Cuba. E-mail: jorge.berlanga@cigb.edu.cu.

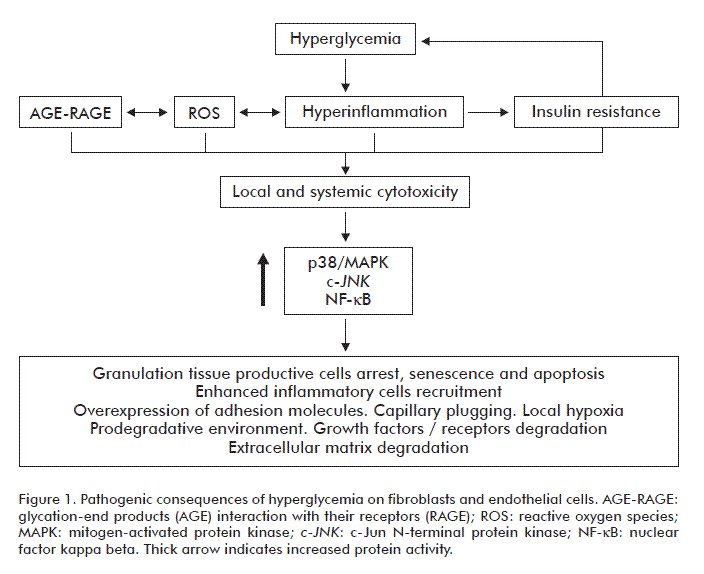{kind=link}
{kind=link}