










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las inmunodeficiencias primarias (IDP) se caracterizan por una susceptibilidad incrementada a infecciones que se debe a defectos genéticos que afectan el desarrollo o función del sistema inmune.1 Con el paso de los años se reconoce que, en dependencia de la enfermedad; la autoinmunidad, autoinflamación, alergia y malignidad pueden manifestarse y, en algunos casos, predominar en el cuadro clínico.
La manifestaciones se han asociado a defectos generalmente monogénicos de la inmunidad producto de mutaciones que resultan en la perdida de expresión, perdida o aumento de función de la proteína codificada.1,2,3 Por este motivo, y para englobar el amplio rango de fenotipos de estos desórdenes, se propuso el actual término de errores innatos de la inmunidad (EII) humana.1,4
Los estudios recientes revelan que las inmunodeficiencias primarias son mucho más frecuentes de lo que se cree, y se estima que alrededor del 1 % de la población tiene alguna forma de inmunodeficiencia primaria cuando se toman en cuenta todas las variantes.5
La susceptibilidad incrementada a procesos infecciosos se considera el principal signo de sugestivo de inmunodeficiencia; sin embargo, otras manifestaciones como las de índole hematológico y/u oncológico pueden ser parte de la evolución e incluso del debut de un error innato de la inmunidad. En la última versión de la clasificación fenotípica de estas enfermedades que establece la Unión Internacional de Sociedades de Inmunología (IUIS) se incorporó un nuevo grupo de entidades nosológicas dentro del IX grupo de errores innatos de la inmunidad: los defectos de la inmunidad por fallo o insuficiencia medular;3 se caracterizan por trastornos oncohematológicos con los consiguientes desperfectos de los mecanismos de defensa.
Algunos errores innatos de la inmunidad poseen predisposición a desarrollar procesos malignos y, aunque el riesgo varía entre ellos, sobresale el peligro de linfoma, leucemia y otras malignidades hematológicas dado por el daño acumulativo de ADN. Los déficits celulares y los defectos de reparación del ADN, seguidos por los déficits de inmunidad humoral son los que con mayor frecuencia evolucionan a una degeneración maligna. Después de las infecciones, algunos estudios reportan que el cáncer es la segunda causa de muerte más común en estos pacientes.6,7
Se describen diferentes trastornos hematológicos en varios errores innatos de la inmunidad; de ellos, al menos 98 pueden cursar con trombocitopenia, 61 con alguna otra citopenia, 59 con anemia, 15 con riesgo de mielodisplasia y 11 con algún tipo de leucemia.8
Es válido sospechar que los pacientes que se atienden por estos trastornos en los servicios de oncohematología y pediatría pudiesen presentar una inmunodeficiencia no diagnosticada y, en tal caso, mal tratada; lo que ensombrece el pronóstico.
Por lo que este estudio se propone como objetivo identificar los pacientes con diagnóstico probable de errores innatos de la inmunidad en el servicio de oncohematología pediátrica del Hospital Pediátrico “Pepe Portilla” de Pinar del Río, en el período de enero de 1979 hasta marzo de 2021.
MÉTODOS
Se realizó un estudio observacional, descriptivo de corte transversal en el servicio de oncohematología del Hospital Pediátrico Provincial Docente “Pepe Portilla” de Pinar del Río. Para ello se revisaron las historias clínicas de los pacientes atendidos en el período de enero de 1979 hasta marzo de 2021.
El universo estuvo constituido por todos los pacientes atendidos en la consulta de oncohematología con edad pediátrica al momento del debut de su enfermedad. La muestra estuvo constituida por un total de 103 pacientes. Para la selección de la misma se tuvieron en cuenta los pacientes con registro e historia clínica confeccionada en dicho servicio.
Se revisaron las historias clínicas de los pacientes muestreados. Se indagaron los antecedentes patológicos personales y familiares para la búsqueda de las siguientes variables: enfermedad oncohematológica diagnosticada, diagnóstico probable de error innato de la inmunidad y signos de alarma de inmunodeficiencia primaria que establece la Fundación Jeffrey Modell,9 en hematopatías no leucémicas y leucémicas.
Se cumplió con los principios de la ética médica y los aspectos establecidos en la Declaración de Helsinki
RESULTADOS
El 87,4 % presentó trastornos oncológicos, donde predominó algún tipo de leucemia (84,5 %). La sobrevida de los pacientes a la edad adulta (>19 años) fue del 65 %. El sexo masculino representó el 45,6 % del total; mientras que el sexo femenino predominó en los pacientes con hematopatías. (Tabla 1)
Tabla 1 Distribución de frecuencias de enfermedades oncohematológicas en el registro de pacientes del servicio de oncohematología del Hospital Pediátrico Provincial Docente “Pepe Portilla” de Pinar del Río según edad y sexo en el período enero, 1979 - marzo, 2021
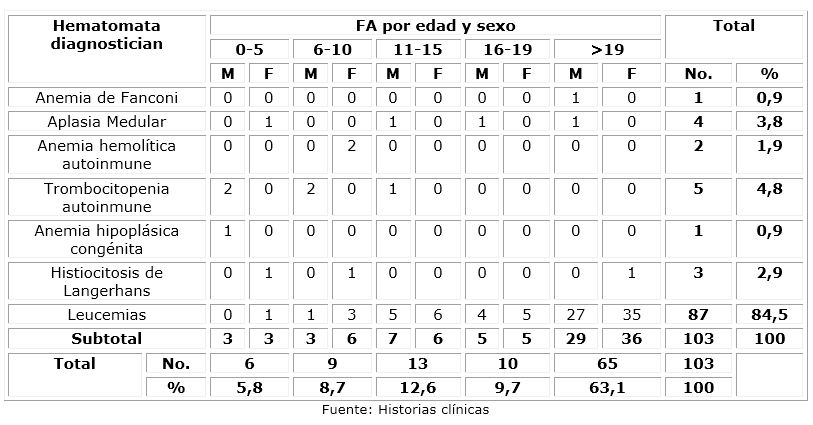
Se hallaron cinco pacientes (4,8 %) con diagnóstico probable de un error innato de la inmunidad: uno con anemia de Fanconi, y cuatro con aplasia medular. (Tabla 2)
Tabla 2 Relación de pacientes con diagnóstico probable de errores innatos de la inmunidad
| Paciente | Error innato de la inmunidad | Grupo | Edad | Sexo |
|---|---|---|---|---|
| 1 | Anemia de Fanconi | IX | 23 | M |
| 2 | Aplasia medular | IX | 25 | M |
| 3 | Aplasia medular | IX | 17 | M |
| 4 | Aplasia medular | IX | 13 | M |
| 5 | Aplasia medular | IX | 4 | F |
Fuente: Historias clínicas
De los 11 pacientes con hematopatías no leucémicas, se constató que seis de ellos (54,5 %) tuvieron al menos uno y hasta tres signos de alarma; solo uno (9,1 %) tuvo más de cuatro y cuatro (36,4 %) no presentaron ningún signo. (Tabla 3)
Tabla 3 Signos de alarma de inmunodeficiencia en pacientes con diagnóstico de hematopatías.
| Diagnóstico | Cantidad de signos de alarma | |||
|---|---|---|---|---|
| Ninguno | Hasta 3 | 4 o más | ||
| Anemia hemolítica autoinmune | 1 | 1 | 0 | |
| Trombocitopenia autoinmune | 2 | 2 | 1 | |
| Anemia hipoplásica congénita | 0 | 1 | 0 | |
| Histiocitosis de Langerhans | 1 | 2 | 0 | |
| Total | No. | 4 | 6 | 1 |
| % | 36,4 | 54,5 | 9,1 | |
Fuente: Historias clínicas
El 72,4 % no presentó ningún signo de alarma y el 24,1 % presentó uno o hasta tres signos. (Tabla 4)

DISCUSIÓN
Conforme a los reportes internacionales, en los pacientes detectados con diagnóstico probable de inmunodeficiencias es más frecuente el sexo masculino.10,11
La anemia de Fanconi se agregó recientemente al repertorio de los errores innatos de la inmunidad humana en el grupo IX de dicha clasificación de inmunodeficiencias por fallo de médula. Esta constituye el trastorno más frecuente del grupo y se asocia al defecto de 21 genes con patrón autosómico recesivo y uno ligado al X.3
El cuadro clínico se caracteriza por pancitopenia en sangre periférica con una médula ósea hipocelular. Se relaciona con frecuencia a otros trastornos congénitos (baja estatura, hipogonadismo, máculas color café con leche, anormalidades de los miembros superiores). Es una de las IDP descrita con fuerte predisposición a degeneración maligna, por defecto de la reparación del ADN.12
La aplasia medular no se describe per se en la clasificación fenotípica de los errores innatos de la inmunidad humana; sin embargo, se observa esta entidad en 12 de estos trastornos.3
La imposibilidad el medio en que se desarrolla la presente investigación de identificar el trastorno molecular asociado y realizar el diagnóstico genético, implica la valoración obligatoria de estos pacientes por el servicio de inmunología, para diagnosticar o descartar en ellos una inmunodeficiencia primaria. Se requiere un tratamiento de apoyo y valoración integral en estos pacientes pues los procesos infecciosos y la degeneración neoplásica son en ellos especialmente flagelantes.7
Las neoplasias que con mayor frecuencia se reportan asociadas a los errores innatos de la inmunidad son los linfomas, particularmente del tipo no Hodgkin.7,13 En el presente estudio no se encuentran pacientes con diagnóstico de linfoma.
Las leucemias mieloides se han asociado a errores innatos de la inmunidad más que las leucemias linfoides, descritas en solo dos enfermedades.3 En la historia clínica de los pacientes diagnosticados con leucemia no se recogen criterios para diagnóstico probable de error innato de la inmunidad.
Los signos de alarma no son criterios diagnósticos definitorios, sino más bien sugerentes; indicadores que permiten evaluar la sospecha diagnóstica de una IDP. Y aunque la fundación Jeffrey Modell,9 y otras sociedades de inmunología clínica han establecido estos y otros criterios a partir de estudios epidemiológicos a gran escala, no se les ha calculado un valor de riesgo, por lo que su asociación es meramente estadística. Sin embargo, es fácil concluir que algunos tienen más “peso” que otros cuando se sospecha una IDP. Por ejemplo, el fallo de medro (o baja talla o bajo peso para la edad), este parámetro se ve afectado en la mayoría sino en todos los niños con enfermedades crónicas o severas;14,15 por lo que no es buen indicador predictor de IDP. No así el antecedente patológico familiar, principalmente de primer grado, ya que al ser en su mayoría enfermedades monogénicas con patrones de herencia conocidos, los errores innatos de la inmunidad pueden ser diagnosticados (diagnóstico probable) con la clínica y la exploración a fondo del árbol genealógico.2,4,13
Se concluye que los signos de alarma de la Fundación Jeffrey Modell para el diagnóstico de inmunodeficiencias primarias no resultaron útiles en los pacientes con hematopatías, por lo que se pudieran implementar otros criterios para la identificación de estos trastornos. Ante tales resultados se impone una reevaluación de estos pacientes por el grupo provincial de inmunología con vistas al diagnóstico de errores innatos de la inmunidad y su tratamiento integral.

