










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
Las inmunodeficiencias primarias (IDP) son un grupo de trastornos frecuentemente graves y a menudo mortales que reflejan un déficit cuantitativo y/o cualitativo en uno o más componentes del sistema inmune. Se caracterizan por susceptibilidad incrementada a infecciones, debida a defectos genéticos que afectan el desarrollo o función del sistema inmune.1 Con el paso de los años se reconoce que, en dependencia de la enfermedad; la autoinmunidad, autoinflamación, alergia y malignidad pueden manifestarse y, en algunos casos, predominar en el cuadro clínico asociado al defecto generalmente monogénico de la inmunidad, producto de mutaciones que resultan en la pérdida de expresión, hipo o hiper función de la proteína codificada.1,2,3 Para englobar el amplio rango de fenotipos de estos desórdenes se propuso el actual término de errores innatos de la inmunidad (EII) humana.1,4
Tradicionalmente se consideraron enfermedades raras, con incidencia de aproximadamente 1 en 10 000 a 50 000 nacimientos. Con la mejor definición de los fenotipos clínicos y el descubrimiento de nuevos errores de la inmunidad se cree que su incidencia general aumenta, al menos, a 1 en cada a 5 000 nacimientos.3 Sin embargo, el número de nuevas inmunodeficiencias ha aumentado con celeridad. Hasta el 2020, se describen más de 430 genes como causas monogénicas de 300 inmunodeficiencias primarias, y estas aumentan con alrededor de 20 nuevas cada año.5,6,7,8
Estudios más recientes revelan que las inmunodeficiencias primarias son mucho más frecuentes de lo que se cree y se estima que alrededor del 1% de la población tiene alguna forma de inmunodeficiencia primaria cuando se toman en cuenta todas las variantes.5
La ausencia o mal función de uno de los componentes del sistema inmune puede determinar la presentación de un EII. A nivel molecular, la mayoría de las IDP están causadas por mutaciones en diferentes genes (heterogeneidad de locus), que por lo general intervienen en algún mecanismo inmunológico, y distintas variantes patogénicas de un mismo locus causan diferentes formas de EII; con frecuencia, aunque no siempre, mediante distintos genotipos (heterogeneidad alélica): lesiones monoalélicas contra bialélicas, variaciones con pérdida (hipomórficas) o ganancia de función (hipermórficas) y dominancia negativa contra haploinsuficiencia.6,9
A partir del entendimiento de las nociones básicas de genética, es comprensible la historia familiar de los pacientes con IDP, y la apropiada interpretación del asesoramiento genético que necesitan, tanto pacientes como familiares en cada caso.6
Generalmente las condiciones más severas comienzan en los primeros meses de vida, lo que causa que los errores innatos de la inmunidad sean confundidos con enfermedades exclusivas de la niñez.2
La anamnesis y evaluación clínica extensivas son las armas que el pediatra posee para sospechar el diagnóstico de un EII, por lo que obtener la información para la historia clínica personal y familiar es de gran importancia. Con frecuencia la identificación de agentes patógenos involucrados en las condiciones infecciosas puede llevar al diagnóstico de alguna de estas afecciones.2
La adquisición de nuevas herramientas diagnósticas asociadas a la biología molecular hace posible incrementar el conocimiento de la genética médica, lo que permite una mejor caracterización de estas enfermedades, más allá de su asociación a las variantes genéticas portadas por los pacientes. El acceso a estas técnicas es aún difícil en Cuba, limitadas a algunos institutos y centros de investigación, de ahí que en atención primaria y secundaria el peso para el diagnóstico de los EII recae en el método clínico.2
La incidencia y prevalencia reales de las inmunodeficiencias primarias es críticamente subestimada, puede distribuirse en diferentes categorías: sobre todo en enfermedades infecciosas, de los órganos hematopoyéticos y neoplasias; debido a diagnósticos inexactos, el compromiso multiorgánico y los múltiples fenotipos de los EII. Por lo que se hace necesario lograr una correcta visión clínica, inmunológica y genética de las IDP en las políticas de salud pública.10
Aunque se acepta que la evaluación diagnóstica de una inmunodeficiencia requiere varios componentes que deberían incluirse en la discusión diagnóstica de un paciente: primero la evaluación clínica, la analítica inmunológica, la evaluación genética y el análisis de estos resultados; luego el seguimiento rutinario, la rediscusión diagnóstica y del tratamiento clínico; no existe una guía clara con respecto a este asunto.2
Otros desafíos relevantes del diagnóstico de inmunodeficiencias primarias en Cuba y América Latina, lo constituyen el desconocimiento por la población y los profesionales de la salud, y la existencia de pocos centros de referencia y de investigación especializados. Esto está muy relacionado con que las inmunodeficiencias primarias aún no son consideradas una prioridad en los sistemas de salud de estos países que deben enfocarse en combatir enfermedades con mayor prevalencia.2
Varios autores plantean la importancia del diagnóstico analítico de los EII, desde los exámenes rutinarios que incluyen la determinación de niveles de inmunoglobulinas y que aumentan en complejidad, luego la citometría de flujo hasta el empleo de técnicas de secuenciación de nueva generación.11
Queda por definir un algoritmo clínico para el desarrollo de los fenotipos en los EII. De ahí, que la presente investigación tenga por objetivo diseñar un modelo para la confección de historia clínica en el paciente con sospecha de error innato de la inmunidad.
METODOS
Se realizó un trabajo de revisión y discusión de los síntomas objetivos y subjetivos a contemplar en la historia clínica de pacientes con errores innatos de la inmunidad según la clasificación de la IUIS, según la experiencia empírica del grupo básico de inmunología en la provincia de Pinar del Río.7
Para ello, se formaron subgrupos de trabajo entre los miembros del grupo provincial de Inmunología de la provincia, en correspondencia con los grupos de clasificación de los errores innatos de la inmunidad, con la posterior discusión colectiva de los aspectos que se agregan a la historia por cada grupo de trabajo.
Se incluye en el presente trabajo las tablas que corresponden a la historia clínica que se propone como resultado de la revisión y discusión.
La estructura que se presenta es dinámica y estará sujeta a revisión periódicamente. El algoritmo para la confección de la propuesta se basa en la historia clínica general de cualquier paciente, y se ajusta a los diferentes fenotipos de los errores innatos de la inmunidad.
RESULTADOS
Consentimiento informado
Se procede a informar mediante documento escrito el objetivo del registro del paciente, que incluye procederes diagnósticos y terapéuticos. Se tiene en cuenta el código de Helsinki. Se solicita la aprobación del paciente o tutor mediante la firma del mismo.
Se toman los datos generales de cada paciente que permitan su identificación y localización.
También se identifica de forma escrita los datos del médico que elabora el documento.

Antecedentes patológicos personales
Una vez reflejado los datos del médico de asistencia se recoge la información referente a los antecedentes patológicos personales.
Se destaca el principal motivo de consulta y a continuación los síntomas relacionados por fenotipos clínicos. En el fenotipo alérgico se declaran siete síntomas, en el infeccioso 39 manifestaciones, en el inflamatorio 38, en cuanto al autoinmune se declaran diez manifestaciones, 52 en el fenotipo que abarca las manifestaciones no inmunológicas y dos en el fenotipo neoplásico.
Se definen los signos de alarma de acuerdo a los síntomas referidos en la historia clínica según los criterios de la Fundación Jefrey Modell.
Se completa este acápite con los antecedentes perinatales, vacunación, factores de riesgo y el orden cronológico de la historia de la enfermedad actual.


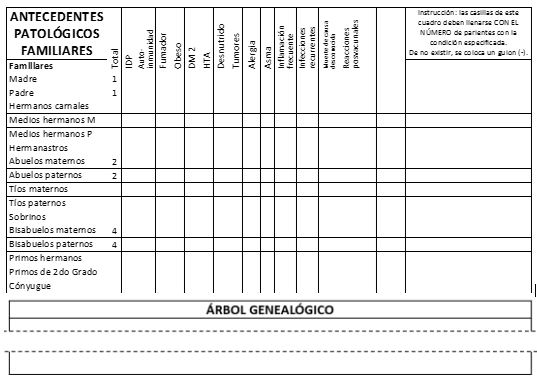
Antecedentes patológicos familiares y árbol genealógico
En los antecedentes patológicos familiares se recopilan los relacionados con el error innato de la inmunidad, enfermedades crónicas, tabaquismo, infecciones y reacciones posvacunales de la familia.
Los antecedentes familiares se resumen en una tabla que especifica el número de familiares por grado y tipo, y la cantidad de estos que se afectan con las entidades señaladas.
A continuación, se elabora el árbol genealógico que da lugar al caso índice.
Examen físico
Se refleja el examen físico general, regional y por sistemas. Se enfatiza en la exploración de la piel, mucosas y faneras, boca y faringe, sistemas respiratorio, cardiovascular, digestivo y hemolinfopoyético.

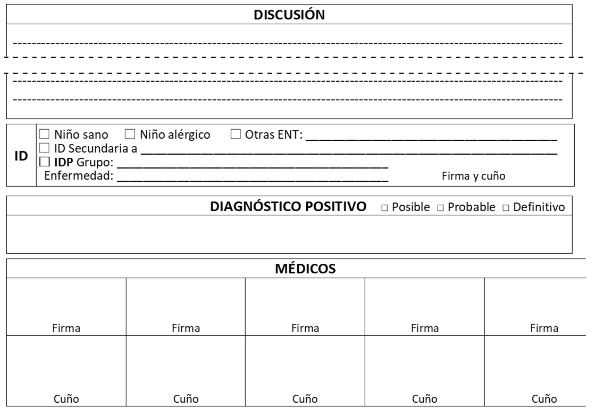
Resumen de los fenotipos clínicos, discusión diagnóstica y clasificación del paciente
Se realiza el resumen de los datos positivos por fenotipos clínicos a partir de la historia clínica. Se procede en el colectivo de la especialidad a realizar la discusión diagnóstica y la clasificación del paciente desde lo general a lo particular, es decir, desde el grupo de inmunodeficiencia hasta el diagnóstico etiológico, de forma definitiva, probable o posible, según corresponda.
DISCUSIÓN
Un aumento de 200 nuevos genes y enfermedades en los últimos 10 años revela un cambio de paradigma de las "inmunodeficiencias primarias" como defectos fundamentales en la respuesta inmune a la infección hacia el concepto más amplio de "errores innatos de la inmunidad", como un grupo integral de diferentes fenotipos, que incluyen alergia, infección, autoinflamación, autoinmunidad y malignidad.12
Es posible que los médicos no puedan reconocer la inmunodeficiencia primaria debido a la rareza de esas enfermedades, particularmente en los países en desarrollo.4 Por tanto, es común que el diagnóstico específico de un error innato de la inmunidad sea retardado, por lo que de forma general se hace difícil para los pediatras y especialistas. De ahí, que la historia clínica debe registrar detalladamente cada uno de los síntomas que corresponden a los diferentes fenotipos de las inmunodeficiencias primarias.13
En cuanto a las manifestaciones alérgicas se reportan tasas más altas de atopia en comparación con la población general en algunas inmunodeficiencias, especialmente aquellas que se deben a deficiencias de anticuerpos. Ello justifica su incorporación en los antecedentes patológicos personales. Por lo tanto, la atopia, en asociación con otros signos bien reconocidos de inmunodeficiencia, debería aumentar la probabilidad diagnóstica de un error innato de la inmunidad subyacente.14
Se destacan entre las características atópicas más comunes la alergia a los alimentos, la dermatitis atópica, el asma, la rinitis alérgica, el aumento de la IgE sérica y la eosinofilia. Estas se superponen con las de los trastornos alérgicos comunes en los pacientes con errores innatos de la inmunidad.14 A medida que se reconocen más errores innatos de la inmunidad, se comienza a apreciar que la inflamación alérgica grave puede ser la única o la manifestación más temprana de defectos inmunitarios monogénicos.15
Las inmunodeficiencias primarias ya no se definen únicamente por la tendencia a las infecciones, pero estas marcan la identificación de estos trastornos. Las infecciones en múltiples localizaciones anatómicas, con aumento de la frecuencia y la gravedad con la edad, se muestran en los errores innatos de la inmunidad.16
De hecho, el aumento de fallecidos por infecciones en estos trastornos quedó evidenciado desde finales del siglo XIX. Sin embargo, los estudios clásicos genéticos revelan que las infecciones graves recurrentes pudieran estar dado por una predisposición genética en el humano. Esta idea crece bajo el sustento de estudios moleculares en tres pasos sucesivos que se solapan. Primero, desde 1985 hacia delante, en los errores innatos de la inmunidad raros subyacen infecciones recurrentes y múltiples con patrón de herencia mendeliano. Segundo, un grupo de infecciones familiares y raras que también se segregan como rasgos mendelianos, pero con resistencia a otras infecciones, se descifraron molecularmente desde los inicios de 1996. Tercero, del 2007 en adelante, un número creciente de infecciones esporádicas comunes o raras, se muestran como resultados de errores innatos monogénicos, pero no mendelianos.17
Según el tipo de infecciones, las infecciones recurrentes y crónicas, particularmente las infecciones pulmonares, son una característica principal de los errores innatos de la inmunidad.4 La infección respiratoria es la complicación más común e incluye sinusitis, otitis aguda, bronquitis, bronquiectasias y neumonía.4
Los inmunodeficientes primarios con complicaciones no infecciosas son cada vez más reconocidos con características de "desregulación inmunitaria" que incluyen autoinmunidad, inflamación, linfoproliferación o malignidad.13
Los primeros signos clínicos o secuelas de las inmunodeficiencias primarias corresponden a la autoinmunidad, tales como citopenias, artritis o enteropatías.13
Un nuevo concepto en los errores innatos de la inmunidad es la disregulación inmune causado por los componentes innatos de la inmunidad en yuxtaposición con la autoinmunidad dirigida por la inmunidad adaptativa. Los trastornos auto inflamatorios son originados por una sobre activación de vías o citocinas proinflamatorias, en su mayoría componentes de los inflamasomas. Estos se manifiestan como fiebre, rash cutáneo, inflamación sistémica, artritis y linfadenopatía.12
Sin embargo, identificar una inmunodeficiencia primaria subyacente en un grupo heterogéneo de pacientes con una variedad de trastornos autoinmunes puede ser una tarea abrumadora. La mayoría de los pediatras o especialistas que atienden a pacientes con trastornos autoinmunes pueden no considerar la evaluación inmunológica en el estudio inicial, ya que genera un subregistro de dichos trastornos. Por lo tanto, no es raro que se retrase el diagnóstico específico de pacientes altamente vulnerables con enfermedad de inmunodeficiencia genética.13
En tal sentido se plantea que, dentro del primer año de presentación clínica, más del 50 % de los pacientes presentaron signos de desregulación inmunitaria y autoinmunidad, especialmente citopenias autoinmunes (21 %), que fue una característica dominante y continuó durante toda su vida.11 Aunque las citopenias autoinmunes a menudo son dominantes en la primera presentación clínica, los pacientes pueden eventualmente progresar a enteropatía autoinmune, enfermedad pulmonar intersticial con o sin granulomas o desarrollar artritis, alopecia y lesiones en el sistema nervioso central.13
El riesgo de malignidades es mayor en pacientes con determinados errores innatos de la inmunidad que en la población general. Sin embargo, la predisposición al tipo de tumor y el entendimiento de los mecanismos celulares y moleculares varía a través de las categorías de los errores innatos de la inmunidad. El defecto molecular per se, que causa el error innato de la inmunidad puede predisponer al origen del tumor. Se sugiere que la malignidad no es consecuencia de la deficiencia inmune, pero ocurre en paralelo a cada uno de estos defectos. Además, en los errores innatos de la inmunidad genéticamente determinados por deficiencias en la reparación del ADN se percibe la predisposición al tumor.18
Adicionalmente, la desregulación de factores epigenéticos relacionados a los errores innatos de la inmunidad o su tratamiento tales como las alteraciones del microbioma pueden contribuir a la predisposición del tumor.18 Se deriva que debe descartarse fumadores pasivos, cuidados diarios, infecciones, anomalías anatómicas e inmunodeficiencia secundaria.19
La anamnesis sobre antecedentes familiares proporciona datos sobre herencia, estilo de vida y ambiente compartidos por toda la familia. El médico de familia debe integrar estos antecedentes médicos familiares junto a otros datos sociológicos y relacionales que constituyen el genograma del paciente. Los antecedentes familiares brindan información necesaria en torno a los antecedentes de errores innatos de la inmunidad o algunos de sus síntomas y/o enfermedades y acerca de la consanguinidad en la familia. Un historial familiar de inmunodeficiencia primaria es el predictor más fuerte de que una persona tenga este tipo de enfermedad. Una evaluación detallada del Reino Unido encontró que las tres señales de advertencia más útiles para la enfermedad de inmunodeficiencia primaria eran antecedentes familiares positivos; un diagnóstico de sepsis tratada con antibióticos intravenosos; y el fracaso para prosperar.16
Por otra parte, una proporción importante de los lactantes y niños afectados por algún error innato de la inmunidad son hijos de matrimonio consanguíneo; donde además, la historia clínica familiar es muy común en estos trastornos por lo que constituye un componente esencial en el diagnóstico.20
Muchas enfermedades de inmunodeficiencia primaria se heredan o se transmiten en las familias. La mayoría se heredan en uno de dos modos diferentes de herencia: recesivo ligado al cromosoma X o autosómico recesivo; rara vez, la herencia es autosómica dominante.21
En la herencia recesiva ligada al X, se pueden encontrar antecedentes familiares de varios varones afectados. La enfermedad se transmite de mujeres (madres) a hombres (hijos). Mientras que los hombres se ven afectados por la enfermedad, las mujeres portadoras generalmente son asintomáticas.21
La enfermedad autosómica recesiva solo ocurre si dos genes anormales (uno de cada padre) están presentes en la descendencia. Hombres y mujeres se ven afectados con igual frecuencia. Ambos padres portan el gen de la enfermedad, aunque ellos son sanos.21
En el raro caso de herencia autosómica dominante, un gen normal no puede compensar la presencia de un gen defectuoso; en esta situación, el gen anormal ejerce un “efecto negativo dominante”.21
El diagnóstico precoz de los errores innatos de la inmunidad es el elemento clave para la reducción de la morbilidad y mortalidad relacionadas con las mismas. El interrogatorio, el examen físico, los antecedentes patológicos personales y familiares, así como la confección adecuada de la historia clínica son elementos imprescindibles para la aproximación a su diagnóstico.

