










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkGaceta Médica Espirituana
versión On-line ISSN 1608-8921
Gac Méd Espirit vol.19 no.1 Sancti Spíritus ene.-abr. 2017
TRABAJO ORIGINAL
Comportamiento de la hipoacusia no sindrómica en una familia del municipio de Urbano Noris. Holguín
Behavior of non-syndromic hearing loss in a family in the municipality of Urbano Noris. Holguín
Dr. Nilson Márquez IbáñezI, Dra. Elayne Esther Santana HernándezII.
I Centro municipal de Genética.Urbano Noris.Holguín.Cuba.
II Centro Provincial de Genética Médica.Holguín.Cuba.
RESUMEN
Fundamento: La hipoacusia hereditaria de causa no sindrómica se presenta frecuentemente en los humanos. Los estudios clínicos y genéticos han permitido conocer mutaciones asociadas a múltiples genes que producen esta enfermedad. Se considera importante saber las características de la presentación de la hipoacusia en cada familia, lo que determinaría su patrón de herencia y así permitiría poder realizar un adecuado asesoramiento genético en estas familias.
Objetivo: Describir las características de la hipoacusia no sindrómica en los afectados de la esta familia estudiada.
Metodología: Se realizó un estudio de serie de casos en una familia del municipio de Urbano Noris, provincia Holguín. El universo de estudio estuvo constituido por los 45 integrantes de la familia, la muestra quedó formada por los 24 enfermos con hipoacusia. Se les solicitó consentimiento informado, se realizó examen físico audiológico a todos los participantes, se logró definir y clasificar los pacientes con el grado de hipoacusia.
Resultados: El grupo de edades que predominó estuvo comprendido entre 16 y 25 años con 7 pacientes; el mayor número de afectados del sexo masculino fue 13 (54,16 %). La hipoacusia moderada se observó en 11 pacientes (45,83 %), seguida por la severa en 8 pacientes (33,33 %). Se determinó la hipoacusia no sindrómica en esta familia con un patrón de herencia autonómica dominante.
Conclusiones: Prevaleció el sexo masculino y la hipoacusia moderada. Se corroboró la gran heterogeneidad clínica descrita para esta enfermedad. Se confirmó la expresividad variable en esta familia.
Palabras clave: Hipoacusia, sordera no asociada a otras enfermedades, deficiencia auditiva, neurofisiología de la audición.
DeCS: PÉRDIDA AUDITIVA/genética; SORDERA/genética.
ABSTRACT
Background: hereditary hearing loss of non-syndromic cause frequently occurs in humans. Clinical and genetic studies have made possible to know mutations associated with multiple genes that produce this disease. It is considered important to know the characteristics of the presentation of hearing loss in each family, determining their inheritance pattern that would allow adequate genetic counseling in these families.
Objective: to describe the characteristics of non-syndromic hearing loss in the affected of this studied family.
Methodology: A retrospective descriptive study was carried out in a family from the municipality of Urbano Noris, Holguín province. The study universe consisted of the 45 members of the family; the sample was formed by the 24 patients with hearing loss. Informed consent was requested, audiological physical examination was performed in all participants, and patients were defined and classified with the degree of hearing loss.
Results: the predominant age group was between 16 and 25 years old with 7 patients, the highest number of males affected, with 13 being (54.16 %). Moderate hearing loss prevailed with 11 patients for (45.83 %) followed by severe hearing loss with 8 patients for (33.33 %).Non-syndromic hearing loss was determined in this family with a dominant autonomic inheritance pattern.
Conclusions: Prevalence of male gender and moderate hearing loss. The great clinical heterogeneity described for this disease was corroborated. The variable expressivity in this family was confirmed.
Keywords: Hearing loss, deafness not associated with other diseases, hearing impairment, hearing neurophysiology.
MeSH: HEARING LOSS/genetics; DEAFNESS/genetics.
INTRODUCCIÓN
La hipoacusia es uno de los trastornos de los sentidos más frecuentes en el ser humano y puede presentarse a cualquier edad. Se calcula que cerca del 10 % de la población adulta muestra algún grado de sordera, y un 33 % de personas mayores de 65 años tienen hipoacusia de magnitud suficiente como para necesitar una prótesis auditiva 1, 2.
De acuerdo con la intensidad de la disminución de la percepción auditiva, pueden ser leves cuando solo surgen problemas de audición con voz baja y ambiente ruidoso; moderadas, cuando se aprecian dificultades con la voz normal, con problemas en la adquisición del lenguaje y en la producción de sonidos; son severas si solo se oyen gritos o se usa amplificación, por lo que no se desarrolla lenguaje a menos que se reciba ayuda 2,3.
También pueden ser profundas, en la que la comprensión es prácticamente nula, incluso con la amplificación, no se llega a producir un desarrollo espontáneo del lenguaje. Al respecto, y en término cuantitativo, se clasifica como leves a aquellas hipoacusias con pérdidas entre 20 y 40 db (decibeles), en el caso de las moderadas el umbral de audibilidad está entre 40 y 60 db, en la categoría severa se aprecian pérdidas entre 60 y 80 db y profundas cuando el umbral está entre 80 a 110 db en cofosis (sordera total) 3-5.
Al momento de producirse la pérdida auditiva, las hipoacusias se clasifican en prelinguales, la lesión se produjo con anterioridad a la adquisición del lenguaje (0-2 años); perilinguales, cuando sucedió durante la etapa de adquisición del lenguaje (2-5 años) y poslinguales cuando la pérdida auditiva es posterior a la estructuración del mismo. Cuanto más precoz aparezca la disfunción auditiva tanto más grave serán sus consecuencias, si se parte del principio que la audición es la vía habitual para adquirir el lenguaje, uno de los más importantes atributos que permiten a los seres humanos la comunicación, que ha tenido una participación decisiva en el desarrollo de la sociedad y sus numerosas culturas 6-8.
Las hipoacusias se pueden también catalogar de acuerdo con la localización en conductivas, perceptivas o neurosensoriales y mixtas. Las hipoacusias genéticas o hereditarias se describen como sindrómicas o no sindrómicas. Las hipoacusias se identifican como sindrómicas cuando se asocian a otros defectos congénitos en órganos o sistemas, puede acompañarse además de malformaciones del oído externo. Por el contrario, las no sindrómicas no se asocian a alguna alteración o defecto de otro sistema, aunque pueden presentar anomalías en el oído medio o interno. Aproximadamente el 30 % de las hipoacusias genéticas prelinguales son sindrómicas, el 70 % restante corresponde a las hipoacusias no sindrómicas. Dentro de las hipoacusias prelinguales no sindrómicas el 80 % se hereda según un patrón autosómico recesivo (AR), el 18 % sigue un patrón autosómico dominante (AD), y el 2 % restante corresponde a las hipoacusias de herencia ligada al cromosoma X y al genoma mitocondrial 9.
Desde el punto de vista estadístico hay estudios realizados que afirman que la hipoacusia es un síntoma frecuentemente diagnosticado tanto en el niño como en el adulto. Censos internacionales han determinado que un 10 % de las poblaciones de Europa y América, presenta una deficiencia auditiva. En relación con la hipoacusia infantil, señala esta estadística que los escolares manifiestan la hipoacusia en más del 13 %. El mismo estudio comprobó que el 50 % de repitentes de grados eran hipoacúsicos 10-12.
El anuario estadístico de salud en Cuba en el año 2009 cita a la sordera entre los primeros 40 padecimientos de la población, ocupando en la distribución porcentual un 2,6 % del total de la población examinada. Según las estadísticas de la Asociación Nacional de Sordos de Cuba (ANSOC), están registrados 14451 sordos e hipoacúsicos, 7830 del sexo masculino y 6621 del sexo femenino, de ellos 1895 son niños.
La provincia de Holguín presenta 2475 afectados con hipoacusia, de estos 2245 son adultos y 230 son niños. Se tiene un estimado que más de la mitad de estos enfermos solo presentan este signo clínico por lo que se encuentran dentro de la hipoacusia no sindrómicas. Existiendo varias familias con múltiples afectados por hipoacusia, lo que constituye un problema de salud en la provincia, por ello se considera necesario desarrollar este estudio con el objetivo de describir características de la hipoacusia no sindrómica en esta familia.
Debido a la existencia de varias familias con múltiples afectados por hipoacusia, lo cual se convierte en un problema de salud en la provincia se hace necesario
MATERIAL Y MÉTODO
Contexto y clasificación del estudio:
Se realizó un estudio tipo reporte de casos en una familia constituida por 45 integrantes, de ellos 24 enfermos con hipoacusia.
Criterios de inclusión:
Hipoacúsicos de esta familia dispuestos a participar en la investigación, con diagnóstico por estudio de potenciales evocados auditivos.
Criterios de exclusión:
Aquellas personas de la familia que no desearon participar y los que después de efectuado el estudio de potenciales evocados auditivos fue normal.
Para el desarrollo de la investigación se respetaron los principios éticos según la Declaración de Helsinki y el establecido por comisión de ética internacional. A todos los integrantes de la familia se les solicitó de forma escrita su aceptación a participar, para ello firmaron el consentimiento informado aquellos que accedieron a asistir voluntariamente y que tenían el derecho de abandonarlo cuando lo consideraran.
Se efectuó revisión de la bibliografía actualizada y de las historias clínicas de los hipoacúsicos ya diagnosticados. Se confeccionó el árbol genealógico y se prosiguió con el examen físico completo y audiológico, se incluyeron potenciales evocados auditivos a todos los integrantes de la familia. Solo 24 presentaban hipoacusia.
Para el procesamiento de la información se creó una base de datos con ayuda del programa microsoft excel con su posterior procesamiento estadístico. La información obtenida se expresó en números absolutos y porcientos, se representó mediante tablas para su mejor comprensión.
RESULTADOS
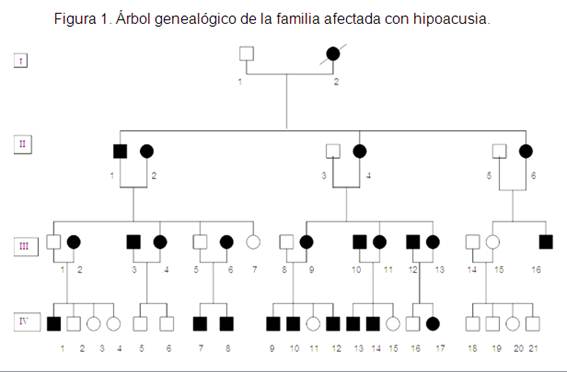
Se presentó una familia con varios afectados con hipoacusia congénita neurosensorial bilateral de grado variable, este signo clínico no progresó con el tiempo, lo cual se comprobó a través de los potenciales evocados auditivos, realizados en un primer momento a los 24 hipoacúsicos diagnosticados y posteriormente al resto con el fin de identificar otros enfermos. Quedó conformado el total de hipoacúsicos en 24, repartidos en las cuatro generaciones. Por la distribución de los afectados en forma vertical, de ambos sexos en todas las generaciones, se planteó un patrón de herencia autosómico dominante.
En la figura 1 se puede apreciar el árbol genealógico de esta familia con sus 45 integrantes de cuatro generaciones, donde a partir de la primera generación aparece una afectada con hipoacusica (ya fallecida), la integrante femenina I-2, que tuvo tres descendientes, de diferentes sexos, afectados pero con diferentes grados de hipoacusia. Como exhibe la tercera generación de nueve descendientes, solo cinco de estos son hipoacúsicos, llama la atención como la pareja conformada por II-1 y II-2 ambos con hipoacusia congénita neurosensorial moderada, solo una hija presentaba la alteración de igual forma que sus padres. En la cuarta generación un solo matrimonio de hipoacúsicos tienen sus dos hijos sanos, los demás afectados sí presentan descendientes enfermos. Estas características confirman el patrón de herencia autosómico dominante, donde la probabilidad de trasmisión a la descendencia es del 50 %.
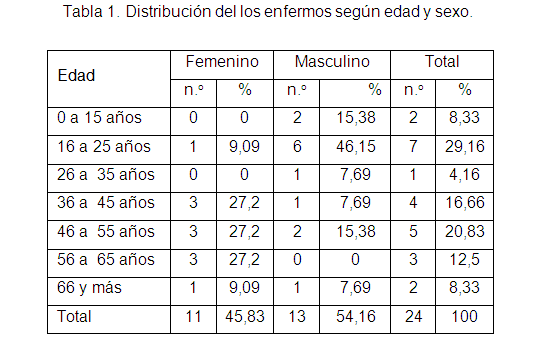
En la tabla 1 se distribuyeron los enfermos según su edad y sexo donde el grupo de edades con mayor número fue el 16 a 25 años con 7 afectados (29,16 %), predominaron los varones con 6 (46,15 %). En total los enfermos masculinos fueron 13 (54,16 %).
La distribución de los afectados se realizó por los mismos grupos de edad, para examinar el grado de hipoacusia y resultó que predominó la hipoacusia moderada con 11 afectados (45,83 %), seguida de la hipoacusia severa con 8 afectados (33,33 %).

DISCUSIÓN
Las hipoacusias neurosensoriales causadas por lesiones en el oído interno o en la vía auditiva nerviosa es la forma más común de déficit auditivo congénito. La incidencia se estima en 1:1.000 recién nacidos. Dentro de las causas conocidas de hipoacusia neurosensorial están las genéticas que son las responsables de más del 50 % de hipoacusias congénitas severas a profundas.
Las hipoacusias no sindrómica se presentan con gran heterogeneidad genética alélica y no alélica de locus, penetrancia reducida y expresividad variable. Se detallan 144 mutaciones diferentes en 22 genes asociados a hipoacusia no sindrómicas 1-3. Esto explicaría el número de enfermos en esta familia algunos de ellos con ambos padres enfermos y otros con padres sanos donde es evidente la penetrancia reducida y como la intensidad también varió en algunos afectados sin guardar relación con lo expresado por sus progenitores.
La prevalencia de la hipoacusia en el recién nacido y el lactante se estima en 1,5 a 6 pacientes cada 1000 nacidos vivos según se trate de severa o de cualquier grado, en la edad escolar, la prevalencia de hipoacusias de más de 45 decibeles es de 3 por cada 1000 nacidos vivos y de cualquier grado hasta 13 por cada 1000 nacidos vivos. En niños con determinados factores de riesgo la incidencia puede elevarse hasta 4 % para hipoacusias severas 4-6.
Se estima que al menos el 60 % de las hipoacusias de inicio precoz responden a una causa genética; el 40 % restante se atribuye a causas ambientales. Entre estas últimas destacan las infecciones prenatales (citomegalovirus, herpesvirus, rubéola, toxoplasma, etc.), las infecciones posnatales (meningitis bacterianas), el sufrimiento fetal, la hiperbilirrubinemia o los fármacos ototóxicos 7. Estos porcientos han ido modificándose por los esfuerzos que realizan el sistema de salud y el gobierno cubano en función de mejorar la calidad de vida, para lograr inclusión adecuada de todos los discapacitados a la vida social. Se ejecutan múltiples investigaciones, gracias a los avances tecnológicos de la biología molecular, que en muchos casos permiten conocer la causa y tratarla siempre que sea posible. Es importante tener en cuenta que la presencia de una causa ambiental no excluye necesariamente la existencia de una predisposición genética subyacente 8,9.
Dentro de las hipoacusias prelinguales no sindrómicas un porciento alto se hereda según un patrón autosómico recesivo, sin embargo actualmente se desconocen los porcentajes correspondientes a cada patrón de herencia 10. Por otra parte, el porcentaje de familias con un patrón autosómico dominante es mayor que en las prelinguales, como lo ocurrido en esta familia con 24 afectados en todas las generaciones.
Por lo complejo que se ha presentado esta enfermedad donde muchos hipoacúsicos han contraído matrimonio con otros enfermos con la misma discapacidad algo que es frecuente en estos casos. Entonces los afectados que presentan la forma más severa la enfermedad pueden tener varias mutaciones. Los pacientes portadores de un alelo mutado en el gen GJB2 nacen hipoacúsicos, pero los tienen esta mutación en homocigosis, habitualmente presentan una hipoacusia severa de inicio precoz 11-12. Esto pudiera explicar lo ocurrido en algunos descendientes como el IV-13 y IV-14 con ambos padres con hipoacusia neurosensorial moderada y ellos tienen una hipoacusia congénita severa.
Si bien el fenotipo suele ser homogéneo, el gen GJB2 es un buen ejemplo de heterogeneidad alélica. Sin embargo se ha comprobado en pacientes portadores de mutaciones en homocigosis que no todos expresan la enfermedad de forma homogénea, ya que el grado de hipoacusia o déficit auditivo se ha mostrado variable (moderado a profundo), e incluso hipoacusias asimétricas. Por otro lado, las mutaciones en GJB2 pueden ser las responsables, tanto de hipoacusias hereditarias no sindrómicas autosómicas dominantes (DFNA3A), como de diferentes síndromes con fenotipos más o menos severosv13,14. Se ha intentado establecer una relación genotipo-fenotipo donde la severidad del déficit se ha correlacionado con el tipo concreto de mutación, obteniendo resultados concluyentes en aquellos estudios con suficiente número de pacientes 15.
En Cuba no contamos con estudios moleculares para llegar a la caracterización molecular de las sorderas autosómicas dominantes, esto dificulta el asesoramiento genético a estas familias. En poblaciones de origen europeo, como esta familia pudiera aparecer una mutación en el gen GJB2 de forma dominante, que se trasmite a sus hijos en un 50 %, pero al contraer matrimonio con otros hipoacusicos con probabilidades de tener otras mutaciones dominantes o recesivas, se producirían combinaciones de varios alelos mutados y es esto explicaría la expresividad variable de la hipoacusia en esta familia con tantos afectados con gran variabilidad en su expresión clinica de la enfermedad 6,8,12.
Se han reportado combinaciones de varias mutaciones dominantes que causan modificaciones en su expresión fenotípica, cuando aparecen como heterocigotos compuestos 13. Esta situación no es habitual pero en familias tan extensas como esta donde los matrimonios fueron quizás escogidos por su condición de sordera pudiera especularse que existen en los descendientes varias mutaciones para que se exprese esta enfermedad de forma diferente.
Por lo frecuente que resulta la hipoacusia neurosensorial congénita no sindrómica, en la provincia se realizan investigaciones para lograr caracterizar clínicamente a esta enfermedad y nos permita así trazar estrategias de estudio, diagnóstico y seguimiento, en función de poder brindar un adecuado asesoramiento genético a cada familia afectada.
CONCLUSIONES
Se determinó la hipoacusia no sindrómica en esta familia con un patrón de herencia autonómica dominante. Se corroboró la gran heterogeneidad genética y clínica de esta enfermedad, se confirmó la expresividad variable de esta afección.
Por lo tanto, en el caso de las hipoacusias genéticas, la penetrancia incompleta, la expresividad variable y la heterogeneidad genética y alélica dificultan el establecimiento de correlaciones entre una determinada mutación (genotipo) y sus manifestaciones clínicas y audiométricas (fenotipo). Este hecho justifica en parte la dificultad para la integración del asesoramiento genético en la práctica clínica habitual, a pesar de los beneficios para los pacientes y sus familiares.
REFERENCIAS BIBLIOGRÁFICAS
1. Vona B, Nanda I, Hofrichter MA, Shehata-Dieler W, Haaf T. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol Cell Probes [Internet]. 2015 Oct [cited: 2016/11/21];29(5):260-70. Available from: http://www.sciencedirect.com/science/article/pii/S0890850815000353
2. Gu X, Sun S, Guo L, Lu X, Mei H, Lai C, et al. Novel biallelic OTOGL mutations in a Chinese family with moderate non-syndromic sensorineural hearing loss. Int J Pediatr Otorhinolaryngol [Internet]. 2015 Jun [cited: 2016/11/21];79(6):817-20. Available from: http://www.sciencedirect.com/science/article/pii/S0165587615001196
3. Iossa S, Costa V, Corvino V, Auletta G, Barruffo L, Cappellani S, et al. Phenotypic and genetic characterization of a family carrying two Xq21.1-21.3 interstitial deletions associated with syndromic hearing loss. Mol Cytogenet [Internet]. 2015 Mar [2016/11/20];8:18. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4376344/
4. Choi HS, Kim AR, Kim SH, Choi BY. Identification of a novel truncation mutation of EYA4 in moderate degree hearing loss by targeted exome sequencing. Eur Arch Otorhinolaryngol [Internet]. 2016 May [2016/11/20];273(5):1123-9. Available from: http://link.springer.com/article/10.1007%2Fs00405-015-3661-2
5. Chen Y, Wang Z, Wang Z, Chen D, Chai Y, Pang X, et al. Targeted next-generation sequencing in uyghur families with non-syndromic sensorineural hearing loss. PLoS One [Internet]. 2015 May 26 [2016/11/20];10(5):e0127879. Available from: http://link.springer.com/article/10.1007%2Fs00405-015-3661-2
6. Liu F, Hu J, Xia W, Hao L, Ma J, Ma D, et al. Exome Sequencing Identifies a Mutation in EYA4 as a Novel Cause of Autosomal Dominant Non-Syndromic Hearing Loss. PLoS One [Internet]. 2015 May 11[2016/11/20];10(5):e0126602. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4427289/
7. Gao X, Su Y, Chen YL, Han MY, Yuan YY, Xu JC, et al. Identification of Two Novel Compound Heterozygous PTPRQ Mutations Associated with Autosomal Recessive Hearing Loss in a Chinese Family. PLoS One [Internet]. 2015 Apr 28[2016/11/20];10(4):e0124757. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4412678/
8. Zhu YM, Li Y, Wang YL, Bian PP, Xu BC, Liu XW, et al. The deafness-causing mutation c.508_511dup in the GJB2 gene and a literature review. Acta Otolaryngol. 2015 Sep [2016/11/20];135(9):914-8. Available from: http://www.tandfonline.com/doi/abs/10.3109/00016489.2015.1035796?journalCode=ioto20
9. Sun Y, Zhang Z, Cheng J, Lu Y, Yang CL, Luo YY, et al. A novel mutation of EYA4 in a large Chinese family with autosomal dominant middle-frequency sensorineural hearing loss by targeted exome sequencing. J Hum Genet [Internet]. 2015 Jun [2016/11/20];60(6):299-304. Available from: http://www.nature.com/jhg/journal/v60/n6/full/jhg201519a.html
10. Thoenes M, Zimmermann U, Ebermann I, Ptok M, Lewis MA, Thiele H, et al. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J Rare Dis [Internet]. 2015 Feb [cited: 2017/10/21];10(1):10-15. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4334766/
11. Dai ZY, Sun BC, Huang SS, Yuan YY, Zhu YH, Su Y, et al. Correlation analysis of phenotype and genotype of GJB2 in patients with non-syndromic hearing loss in China. Gene. 2015 Oct [cited: 2017/10/21];570(2):272-6. Available from: http://www.sciencedirect.com/science/article/pii/S0378111915007
12. Mittal R, Patel K, Mittal J, Chan B, Yan D, Grati M, et al. Association of PRPS1 Mutations with Disease Phenotypes. Dis Markers [Internet]. 2015 [cited: 2015/11/19];2015:127013. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26089585.
13. Kim J, Jung J, Lee MG, Choi JY, Lee KA. Non-syndromic hearing loss caused by the dominant cis mutation R75Q with the recessive mutation V37I of the GJB2 (Connexin 26) gene. Exp Mol Med [Internet]. 2015 Jun [cited: 2015/11/19];47:e169. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4491724
14. Wasano K, Mutai H, Obuchi C, Masuda S, Matsunaga T. A novel frameshift mutation in KCNQ4 in a family with autosomal recessive non-syndromic hearing loss. Biochem Biophys Res Commun [Internet]. 2015 Aug 7 [cited: 2016/11/ 31]; 463(4):582-6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26036578.
15. Mizutari K, Mutai H, Namba K, Miyanaga Y, Nakano A, Arimoto Y, et al. High prevalence of CDH23 mutations in patients with congenital high-frequency sporadic or recessively inherited hearing loss. Orphanet J Rare Dis [Internet]. 2015 May [2015/11/13];10:60. Available from: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4451718/
Recibido: 2015-09-17
Aprobado: 2017-02-24
Dr.Nilson Márquez Ibáñez. Centro municipal de Genética. Urbano Noris. Holguín. Cuba.

