Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Médica Electrónica
versión On-line ISSN 1684-1824
Rev.Med.Electrón. vol.40 no.2 Matanzas mar.-abr. 2018
PRESENTACIÓN DE CASOS
Síndrome de Sézary. Presentación de un caso
Sézary síndrome. Case presentation
Dra. Yisel Piña Rodríguez, Dr. José Jorge Piña Russinyol, Dra. Diana M. Hernández Fernández, Dra. Meilyn Fernández Martori, Dra. Cristy Darias Domínguez
Hospital Universitario Clínico Quirúrgico Provincial Comandante Faustino Pérez Hernández. Matanzas, Cuba.
RESUMEN
El síndrome de Sézary constituye la fase leucémica de la micosis fungoide caracterizado por eritrodermia, adenopatías superficiales y células atípicas en sangre. Predomina en los hombres con una proporción 2/1 respecto a las mujeres, y en las edades entre los 60 y 70 años de edad. La enfermedad es de difícil tratamiento, con un pronóstico reservado por su baja supervivencia. Por ser infrecuente y su posible similitud con otras dermatosis, se presenta un caso con antecedentes de psoriasis vulgar con 5 años de evolución, que hacía aproximadamente 6 meses, se encontraba sin mejoría en brote de agudización a pesar de los tratamientos indicados.
Palabras clave: síndrome de Sézary, psoriasis, caso infrecuente.
ABSTRACT
Sezary syndrome is the leukemic part of the fungoid mycosis, characterized by erythroderma, surface adenopathies and atypical cells in blood. It predominates in men with a 2/1 proportion in respect to women, and in ages ranging from 60 to 70 years. It is a difficult treated disease, with a reserved prognosis because of the low survival. Due to its infrequency and possible similarity to other dermatosis, it is presented a case with antecedents of vulgar psoriasis of 5 years evolution, without improvement for around 6 months, in acute outbreak in spite of the indicated treatments.
Key words: Sezary syndrome, psoriasis, infrequent case.
INTRODUCCIÓN
La micosis fungoide al igual que el síndrome de Sézary constituyen las variantes más representativitas y de mayor incidencia de los linfomas cutáneos de células T (LCCT). Ocupan el 25 % de todos los linfomas cutáneos y tienen implícitos una amplia gama de procesos linfocíticos malignos con afinidad por la piel, preferentemente por la epidermis.
El síndrome de Sézary constituye la fase leucémica de la micosis fungoide, caracterizado por eritrodermia, poliadenopatías superficiales; y un elevado número de linfocitos atípicos, (células de Sézary) en sangre periférica. Predomina en los hombres con una proporción 2/1 respecto a las mujeres, afectando a las edades comprendidas entre 60 y 70 años.1
Su forma clásica, suele comenzar de novo o precedido de alguna dermatosis inespecífica. Los pacientes pueden presentar todos los componentes del síndrome o sólo uno, el más común, la eritrodermia y desarrollar otros después. Generalmente muestra una evolución más rápida y agresiva que la micosis fungoide, pero una vez que estén comprometidos los ganglios linfáticos y órganos internos puede tener un desenlace rápido y fatal.2
Actualmente se constata que esta enfermedad aumenta su incidencia, mientras que la etiología de la misma permanece desconocida, pues no son concluyentes estudios que tratan de relacionarlo con factores ambientales y con agentes infecciosos como el aislamiento del virus HTLV-1(virus linfotrópico de células humanas, perteneciente a la familia de los oncovirus).
Tampoco es concluyente la asociación con otros virus de la familia herpes viridae como el citomegalovirus (CMV) y el virus de Epstein Barr (EBV). Igualmente sucede con aquellos que lo relacionan con anomalías cromosómicas; debido a deleciones y traslocaciones en los cromosomas 1 y/o 6, así como asociaciones con antígenos de histocompatibilidad, tales como: AW31 y AW32, B8, BW 38 y DR5. Se conoce que todos estos factores son capaces de generar una oncogénesis inmunológica frente a antígenos desconocidos, pero los mismos aún no han sido totalmente definidos.3-5
Habitualmente las lesiones cutáneas comienzan con características inespecíficas, pasan por estado en placas, después tumoral y en ocasiones eritrodermia, con posterior manifestación extracutánea o no. La manifestación extracutánea inicial a menudo es en los ganglios linfáticos.
Es preciso destacar que actualmente se describen múltiples variantes, atípicas, muy diferentes en su presentación clínica a las formas clásicas ya descritas; lo que establecen la denominación de gran simuladora para esta entidad por su estrecha semejanza con múltiples dermatosis benignas que tienen como característica la evolución tórpida con rebeldía a tratamientos convencionales. Es por ello que se debe ampliar el pensamiento clínico de cada facultativo y plantear esta posibilidad diagnóstica.
En las formas típicas el diagnóstico resulta muy fácil tanto desde el punto de vista clínico como histológico, aún así los autores consideran necesaria la realización de técnicas especiales para su corroboración.
Teniendo en cuenta la dificultad diagnóstica que en ocasiones ofrece esta enfermedad, en sus etapas iniciales, dada su similitud con otras dermatosis, se presenta un paciente con síndrome de Sézary en el cual los exámenes de laboratorio y la histopatología apoyaron el diagnostico.
PRESENTACIÓN DEL CASO
Motivo de consulta: fiebre, picazón y escamas en todo el cuerpo.
Centro de asistencia: Hospital “Comandante Faustino Pérez Hernández”. Matanzas, Cuba.
Historia de enfermedad actual: paciente masculino, de raza blanca, con 69 años de edad. Procedente del municipio de Varadero en la provincia de Matanzas, con antecedentes de psoriasis vulgar desde hace 5 años; la cual nunca se corroboró por biopsia de piel, aplicando para ello diversos tratamientos tópicos sin lograr mejoría de las lesiones. Por lo anterior el paciente es ingresado en la institución, ya que tres semanas antes del ingreso, las lesiones de psoriasis comenzaron a empeorar. Aparecieron nuevas lesiones en placas que paulatinamente aumentaron de tamaño, se generalizaron, enrojeciéndose toda la piel y agravándose el prurito. Además apareció fiebre de hasta 39ºC, astenia y aumento de volumen en ambas zonas axilares. Con este cuadro clínico es remitido de su área de salud hacia atención secundaria, Servicio Provincial de Dermatología, del mencionado hospital con el diagnostico de eritrodermia psoriática.
Antecedentes personales del paciente: psoriasis vulgar.
Antecedentes personales y familiares: no refiere.
Examen físico general
Fascie: ectropión
Hemolinfopoyético: adenopatías palpables y visibles, grandes fibroelásticas, movibles, no dolorosas al tacto, de distribución localizada en cadenas cervical, supraclavicular, axilar e inguinales.
TCS: infiltrado en ambos tobillos, de fácil Godet, ligeramente dolorosos a la palpación.
Examen dermatológico
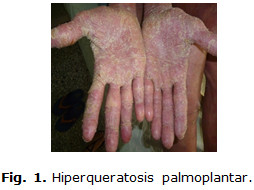
Piel: lesiones eritematoescamosas que confluyen formando grandes placas; algunas infiltradas, otras planas, cubiertas en su superficie por escamas blanquecinas, secas y gruesas. Se desprendían con facilidad, de distribución universal, presentaban marcada hiperqueratosis palmoplantar, así como engrosamiento y pérdida del lustre de las 20 láminas ungueales (onicodistrofia). Figura 1
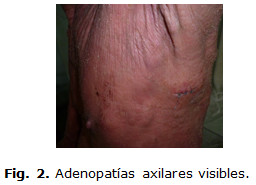
Lesiones tumorales redondeadas, violáceas, de superficie lisa, brillante, no dolorosas a la palpación; que oscilaban entre 1-2 cm de diámetro de distribución localizada en caras laterales y posteriores de ambas piernas. Presentaba eritrodermia generalizada y adenopatías axilares visibles, se constata herida quirúrgica resultado de incisión para biopsia de piel y ganglio. Fig. 2
Complementarios indicados
Leucograma: 20x10L. stab0.01.
Segmentados: 0.52.
Linfo: 0.15.
mono: 0.04.
eos: 0.03.
Linf Cerebriformes 25 %
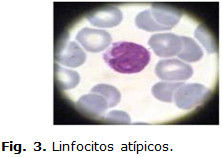
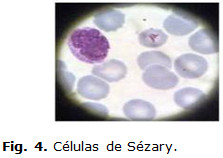
Lámina periférica: normocromía, normocitosis, plaquetas adecuadas en número, leucocitosis con presencia de células linfoides atípicas con núcleos cerebriformes. Aisladas células jóvenes del gránulo en periferia. Figura 3 y Figura 4
- Proteínas totales: 80.9
- Cituria: proteínas: ligeras trazas, leucocitos 100 000, hematíes 10 000, cilindros.
- Electrocardiograma: normal.
- Rayos X de tórax: No se observaron alteraciones pleuropulmonares.
- Ultrasonido abdominal: Sin alteraciones.
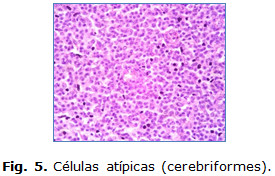
- Biopsia de piel y de ganglio linfático axilar: informa infiltración nodal casi total, por linfocitos neoplásicos grandes y medianos de contorno irregular con mitosis frecuente. Cambios dermatopáticos en la cortical del ganglio. Estadio clínico III. Inmunohistoquimica: CD4 +. Se observa linfocitosis difusa, células atípicas (cerebriformes). Figura 5
-
Se concluyó el caso con un diagnóstico de síndrome de Sézary. Estadio: T4N3B1M1.
Se orientó como tratamiento con prednisona 30 mg/d y crema esteroidea (triamcinolona 2 v/d). Mejoró el cuadro de la eritrodermia y las manifestaciones generales y se decide remitir a Hospital “Hermanos Ameijeiras” para posterior tratamiento específico de la entidad. El paciente se niega y comienza tratamiento por parte de Servicio de Oncología.
Interferón alfa recombinante: 3 millones 3V/ semana.
Quimioterapia (COP): ciclofosfamida 650mg/m2, vincristina 1,4mg/m2 y prednisona 40mg/m2.
Transcurrido 3 meses de tratamiento oncológico el paciente comienza a presentar síntomas de insuficiencia respiratoria, quemando etapas de manera acelerada, lo que provocó el fallecimiento el 19 de diciembre del 2012.DISCUSIÓN
El síndrome de Sézary, a pesar de mostrar tendencia al incremento en la actualidad, continúa siendo una forma rara e infrecuente de linfoma cutáneo de células T (LCCT). Se caracteriza por eritrodermia, compromiso hematológico precoz, curso clínico agresivo y principalmente mal pronóstico.
El diagnóstico inicial sigue siendo difícil, sin embargo los adelantos en el campo de la inmunogenética han hecho posible un avance extraordinario en el conocimiento de esta enfermedad, y a partir de ellos se han propuesto criterios concretos que propician a que se pueda diagnósticar con certeramente. Cuando se detecta la presencia de 1 000 células de Sézary circulantes por mm3, relación CD4/CD8 > 10, expresión aberrante de marcadores pan T, evidencias de células T clonales o un clon de células T con anormalidades cromosómicas.5,6
Además, para considerar el pronóstico y orientar tratamiento deberá estadiarse el paciente según la clasificación TNBM para los Linfomas o Sistema de estadiaje adoptado por Bun y Lamberg en 1979 del National Cancer Institute (NCI) y que se mantiene vigente en la actualidad.
T = Afectación cutánea
T1 Placas limitadas afectación – 10 %.
T2 Placas generalizadas afectación + 10 %
T3 Tumores cutáneos.
T4 Eritrodermia.N= Afectación Ganglionar
No afectación ganglionar.
N1 Ganglios palpables histológicamente negativos.
N2 Ganglios no palpables histología positiva.
N3 Ganglios palpables histológicamente positivos.B= Sangre Periférica
B0 Células de Sézary - 5 %
B1 Células de Sézary + 5 %M= Afectación visceral
M0 No afectación visceral.
M1 Afectación visceral.Según los estudios de Benner,4 sobre pronóstico de supervivencia en pacientes con micosis fungoide-síndrome de Sézary, en el estadio IA, existen tasas aproximadas a 100 % de 5 y 10 años, idénticas a las observadas en adultos sanos de la misma edad, sexo y raza. Según avanza la enfermedad estas descienden en relación a la población sana de la misma edad, hasta llegar al 40 % de supervivencia a los 5 años en enfermedad tumoral y eritrodérmica (T3 y T4); si existe con enfermedad visceral la supervivencia es de 6 a 8 meses. En el síndrome de Sézary las tasas son 11 % a los 5 años. Se deduce que el estadio clínico es el factor fundamental que define el pronóstico de la enfermedad.
Múltiples modalidades terapéuticas han sido utilizadas en el tratamiento de la misma, aunque hasta el momento no existe tratamiento curativo para estos linfomas. Después de determinar el estadio clínico del paciente se orientará la terapéutica requerida. Se han utilizado terapias dirigidas únicamente a la piel, terapias sistémicas y terapias físicas. Se recomienda que los tratamientos deban ser escalonados, iniciando medidas con mejor perfil de seguridad hasta llegar a aquellos tratamientos más agresivos solo en caso de resistencia o enfermedad generalizada.6-9
Por lo tanto, si se toma como punto de partida que el estadio clínico es el factor fundamental que define el pronóstico de la enfermedad y la incidencia de la misma es baja, en relación con otras dermatosis, aparejado a su semejanza en sus estadios iniciales con enfermedades dermatológicas frecuentes que no ofrecen compromiso para la vida del paciente.
Los autores consideran que se impone que ante una enfermedad cutánea aparentemente común, que no mejore con la terapéutica establecida en el tiempo promedio, se indique estudio histopatológico oportuno para diagnosticarla y evitar desenlaces tan efímeros y fatales como el que se expuso en esta ocasión.
REFERENCIAS BIBLIOGRÁFICAS
1- Olsen EA, Rook AH, Zic J, et al. Sézary syndrome: immunopathogenesis, literature review of therapeutic options, and recommendations for therapy by the United States Cutaneous Lymphoma Consortium (USCLC). J Am Acad Dermatol. 2011 Feb;64(2):352-404. Citado en Pubmed; PMID: 21145619.
2- Fitz Patrick TB .Dermatología en Medicina General. T. 3. 7ma ed [Internet]. Argentina: Editorial Médica Panamericana; 2015 [citado 16 Nov 2016]. Disponible en: https://www.agapea.com/libros/Fitzpatrick-Dermatologia-en-Medicina-General-7-edicion-Tomo-3-9789500617024-i.htm3- Alvarado Segura CA, Collazo Caballero SE, Hidalgo González D. Estudio retrospectivo de Micosis Fungoide en un periodo de cinco años en el Hospital Hermanos Ameijeiras. Rev Folia Dermatológica Cubana. 2012 [citado 16 Nov 2016];6(3). Disponible en: http://bvs.sld.cu/revistas/fdc/vol6_3_12/fdc05312.htm
4- Benner MF, Jansen PM, Vermeer MH, et al. Prognostic factors in transformed mycosis fungoides: a retrospective analysis of 100 cases. Blood. 2012 Feb 16;119(7):1643-9. Citado en Pubmed; PMID: 22160616.
5- Quaglino P, Pimpinelli N, Berti E, et al. Time course, clinical pathways, and long-term hazards risk trends of disease progression in patients with classic mycosis fungoides: a multicenter, retrospective follow-up study from the Italian Group of Cutaneous Lymphomas. Cance. 2012;118(23):5830-9. Citado en Pubmed; PMID: 22674564.
6- Olsen EA, Whittaker S, Kim YH, et al. Clinical end points and response criteria in mycosis fungoides and Sézary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. J Clin Oncol. 2011 Jun 20;29(18):2598-607. Citado en Pubmed; PMID: 21576639.
7- Kadin ME, Hughey LC, Wood GS. Large-cell transformation of mycosis fungoides-differential diagnosis with implications for clinical management: a consensus statement of the US Cutaneous Lymphoma Consortium. J Am Acad Dermatol. 2014 Feb;70(2):374-6. Citado en Pubmed; PMID: 24438952.
8- Hughes CF, Khot A, Mc Cormack C, et al. Lack of durable disease control with chemotherapy for mycosis fungoides and Sézary syndrome: a comparative study of systemic therapy. Blood. 2015 Jan 1;25(1):71-81. Citado en Pubmed; PMID: 25336628.
9- Kubica AW, Davis MD, Weaver AL, et al. Sézary syndrome: a study of 176 patients at Mayo Clinic. J Am Acad Dermatol. 2012 Dec;67(6):1189-99. Citado en Pubmed; PMID: 22640839.
Recibido: 18/12/16
Aprobado: 6/12/17Yisel Piña Rodríguez. Hospital Universitario Clínico Quirúrgico Provincial “Comandante Faustino Pérez Hernández”. Carretera central km 101. Matanzas, Cuba.Correo electrónico: jjrussinyol.mtz@infomed.sld.cu