










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La hiperplasia suprarrenal congénita (HSC) es una enfermedad autosómica recesiva caracterizada por un déficit del funcionamiento de una de las enzimas implicadas en la biosíntesis de cortisol a partir del colesterol. (1-3
Constituye la causa más frecuente de ambigüedad sexual, ambos sexos son afectados por igual y las manifestaciones clínicas dependen del déficit enzimático en cuestión, así como, de la etapa de la vida en la que se presenten. (4) Aproximadamente el 95 % de los casos se producen por mutaciones en el gen CYP21A2 localizado en el cromosoma 6p21.3, el cual codifica la enzima 21-OH. (5-8
Los resultados de diferentes programas de tamizaje en el mundo, demuestran que la HSC es relativamente común. (9,10
La prevalencia estimada a nivel mundial oscila de 1:10 000 a 1: 20 000, y la incidencia anual se estima que está entre 1: 5 000 a 1: 15 000. (11)
Desde el punto de vista clínico el déficit de 21-OH se caracteriza por presentar un espectro muy amplio de manifestaciones, las cuales constituyen el resultado de la afectación genética molecular, es decir, esta heterogeneidad clínica está directamente ligada al grado de afectación de los alelos del gen CYP21A2. (3,6,12,13
Se clasifica en 2 grandes grupos: formas clásicas y no clásicas y se considera un tercer grupo que incluye las formas crípticas. Dentro de las formas clásicas se incluye la variedad perdedora sal (PS) y la virilizante simple (VS). Las formas no clásicas se manifiestan en la infancia por la presencia de pubarquia precoz, seudopubertad precoz e incluso una pubertad precoz central; en la adolescencia se muestra con manifestaciones de hiperandrogenismo, dadas por grados variables de acné, hirsutismo, trastornos menstruales, ovulatorios; y de la fertilidad, en la adultez. Las formas crípticas, por su parte, son asintomáticas y solo detectadas en estudios poblacionales. (2,3,6,14
En los últimos años el pronóstico de la HSC ha mejorado sustancialmente a partir de la implementación del tamizaje neonatal en la población general, lo que hace posible la identificación de casos en etapas más tempranas de la vida, debido a la incorporación de nuevas tecnologías que permiten acortar los tiempos al diagnóstico y tratamiento, con la consecuente mejora en el pronóstico de los casos detectados y confirmados. (11)
En los pacientes diagnosticados por el programa cubano de detección precoz debe realizarse el diagnóstico mediante un análisis genético-molecular. Por ello, se diseñó esta investigación con el objetivo de identificar la mutación I172N en los pacientes con diagnóstico de HSC por deficiencia de 21-OH; delinear el fenotipo clínico a partir de los genotipos determinados, que permita realizar un acercamiento a la relación genotipo-fenotipo de esta entidad en la población cubana. Además de complementar el diagnóstico bioquímico neonatal con un estudio molecular más amplio, que incluya a la mutación, que según estudios de correlación genotipo-fenotipo realizados en diferentes regiones del mundo, se describe como la más relacionada con la forma clásica virilizante simple. También se elevaría la calidad del asesoramiento genético prenatal y posnatal de individuos afectados y de parejas de alto riesgo de tener descendencia afectada.
Métodos
Se realizó un estudio de tipo descriptivo, de corte transversal, en pacientes cubanos con diagnóstico de HSC por déficit de 21-OH atendidos en el Instituto de Endocrinología de la Habana que abarcó el periodo comprendido entre 2014-2016. El universo de estudio quedó constituido por 32 pacientes. La información se recogió mediante consulta de las historias clínicas de los pacientes con HSC por déficit de 21-OH y los datos recogidos fueron llevados a una planilla diseñada al efecto. Las variables analizadas fueron: edad, sexo social, edad al diagnóstico, forma clínica de HSC, diagnóstico por programa de pesquisa, antecedentes familiares de HSC, consanguinidad, muerte neonatal inespecífica, crisis genital del recién nacido, vómitos, pérdida de peso, episodios diarreicos, alteraciones electrolíticas, genitales ambiguos, hiperpigmentación genital, macrogenitosomía, trastornos menstruales, hirsutismo, pubertad precoz, pubarquia, acné, virilización de los genitales externos, cromatina oral, sexo cromosómico, gen SRY, diagnóstico molecular previo, mutaciones estudiadas con anterioridad, resultado de estudio molecular anterior, mutación I172N gen CYP21A.
Se tuvieron en cuenta los principios bioéticos de la investigación, por lo que este estudio fue aprobado por el consejo científico y de ética de la institución. Para la identificación de la mutación I172N en el gen CYP21A2 se realizó el proceso de estandarización, según el protocolo llevado a cabo por Plensa Nebot y cols. (15) aplicado en las condiciones del estudio. Los resultados del análisis molecular fueron evaluados y de acuerdo a la forma de distribución de los datos se aplicaron test estadísticos descriptivos con medidas de tendencia central como promedio, razones y test de asociación Chi2 y se consideraron los resultados significativos para una p≤0,05. El programa de análisis que se utilizó fue SPSS v 19,0 y los resultados se presentaron en tablas o gráficos según fue más factible mostrar la información.
Resultados
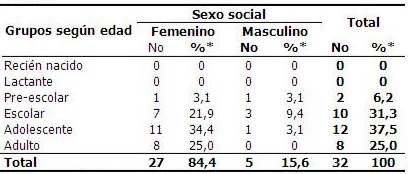
Respecto a la distribución de los pacientes estudiados de acuerdo a grupos de edades y sexo predominó el sexo femenino con 27 pacientes que representó el 84,4 % y solo 5 (15,6 %) correspondieron al sexo masculino, para una razón F: M de 5:1; lo cual está en correspondencia con lo que se esperaría para los rasgos influidos por el sexo. En los varones evaluados predominaron los que se encontraban en edad escolar (9,4 %) y en su mayoría, las pacientes femeninas estudiadas fueron adolescentes (34 %). (Tabla 1).
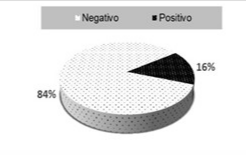
De los 32 pacientes estudiados no fue posible detectar la mutación en un 84 % de los casos, solo 5 (16 %) resultaron positivos para la mutación objeto de estudio. (Gráfico 1).
Tres de las muestras analizadas mostraron 2 bandas; una de 416pb correspondiente al alelo normal y otra de 394pb correspondiente al alelo mutado, resultaron ser heterocigóticos para la mutación y en otras 2 muestras se pudo identificar, una única banda correspondiente a 394pb; resultaron estos pacientes genotípicamente homocigóticos. (Figura 1).
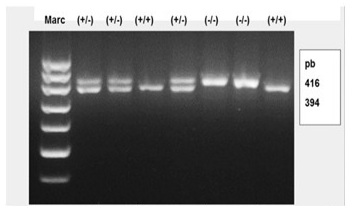
Fig. 1 - Foto de un gel de agarosa al 3 % donde se muestra la corrida electroforética de los productos de PCR.
Al analizar la distribución de la población estudiada en cuanto a sexo y genotipo para la mutación I172N se encontró, que del total de casos positivos (n=5) el 80 % pertenecían al sexo femenino, presentándose tanto en homocigosis como en heterocigosis. El único varón afectado resultó ser genotípicamente heterocigótico. (Tabla 2).

Se estimó la frecuencia de la mutación I172N en los pacientes con HSC por déficit 21-OH, se consideró solo la población de pacientes estudiados. De los 64 alelos pesquisados, 7 presentaron la mutación (2 individuos homocigóticos y 3 heterocigóticos), lo cual corresponde con una frecuencia de 10,9 %. Se logró determinar en la población estudiada (n=32), las frecuencias de las mutaciones P30L, IVS2-12A/C-G (Intrón2), del 8pb y G318X y al realizar un análisis comparativo entre ellas, se encontró que la mutación intrón 2 se presentó como la de mayor frecuencia con un 21,8 %, seguida de G318X e I172N, ambas con frecuencia de 10,9 %, por su parte, P30L y del 8pb, mostraron a su vez igual frecuencia con 4,6 %. (Gráfico 2).

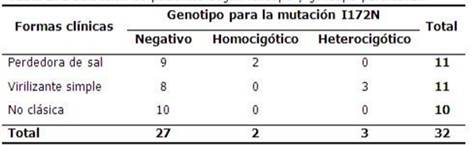
Se muestra que en esta población no se encontró ningún caso asociado a las formas no clásicas, se observó que la mutación fue detectada en pacientes con formas clínicas clásicas, o sea, todos los casos que resultaron heterocigóticos se presentaron con un fenotipo clínico virilizante simple y los casos positivos genotípicamente homocigóticos para I172N, se relacionaron con la expresión más grave de la enfermedad, forma perdedora de sal. Se comprobó con estos resultados que estadísticamente los genotipos determinados y las formas clínicas encontradas estaban significativamente asociados. (x2. p=0.04, p≤0.05). (Tabla 3).
Al analizar de manera individualizada los fenotipos esperados y los observados en los pacientes que resultaron afectados con la mutación objeto de estudio se pudo encontrar que los que tuvieron el genotipo homocigótico, no se presentaron con la forma virilizante simple como se esperaba. El individuo genotípicamente I172N/I172N, se diagnosticó como una forma perdedora de sal, al igual que el paciente con el genotipo más grave (P30L, I2, del 8pb, I172N/I172N). (Tabla 4).

Se muestran algunos datos clínicos de interés de cada uno de los casos en los que fue posible detectar la mutación I172N, se analizó teniendo en cuenta el sexo y el genotipo en cada caso. Cabe señalar, que solo se identificó un paciente del sexo masculino con la mutación y con fenotipo clínico virilizante simple. Por su parte las féminas se presentaron en un 50 % con el fenotipo virilizante simple y el otro 50 % con la variedad perdedora de sal. El genotipo G318X en heterocigosis compuesta con la mutación objeto de estudio (I172N) se presentó en ambos sexos. Otro de los casos analizados se presentó con la forma virilizante simple, sexo femenino y genotípicamente heterocigótico para I172N, igual que la anterior, pero con la mutación I2 en uno de sus alelos, lo que lo convierte también, en heterocigótico compuesto. Se pudo encontrar clínicamente como dato significativo, una paciente que no fue diagnosticada por el programa, pues al momento de su nacimiento aún no se contaba con la pesquisa como una herramienta para la detección precoz en los neonatos afectados con esta patología. Presentó crisis genital del recién nacido, pero no se encontró virilización genital, ni clitoromegalia; solo se recoge como dato positivo la pubertad precoz, por lo que no procedía realizar ningún estudio para la determinación de sexo en este caso. Es necesario aclarar, que se encontró positividad en todos los estudios hormonales realizados, que la ubican en esta forma clínica virilizante simple y no en una forma no clásica de la enfermedad, a pesar de no presentar signos de virilización genital al momento del diagnóstico.
Se tuvo 2 pacientes en los que la forma perdedora de sal se presentó asociada al genotipo homocigótico para la mutación I172N ambos del sexo femenino y fueron diagnosticados entre 7-15 días de nacido, uno por programa y el otro no, dado que en este último no estaba implementado el programa de pesquisa aún.
Las manifestaciones clínicas del paciente heterocigótico compuesto para P30L, I2 y del 8pb (genotipo más grave), no se presentó con toda la clínica de una pérdida salina, solo tuvo al momento del diagnóstico alteraciones electrolíticas (hiponatremia e hiperpotasemia). La virilización genital requirió un procedimiento quirúrgico (abocamiento de la vagina) y la hipertrofia del clítoris fue leve (estadío Prader 1), con esta clínica se le indicaron estudios cromosómicos y moleculares para diagnóstico de sexo y resultó cromosómicamente femenina (46, XX).
El otro caso con fenotipo perdedor de sal y único con genotipo homocigótico para I172N, sin otras mutaciones detectadas hasta el momento, de las que se pesquisan de rutina para esta enfermedad, sí se presentó con toda una clínica florida de la variedad perdedora de sal: vómitos, diarreas, descompensación hidroelectrolítica, genitales ambiguos (estadío Prader 2), entre otras. (Tabla 5).

Discusión
La HSC puede estar sujeta a muchas dificultades diagnósticas, ya que su cortejo sintomático es parecido al de otros errores congénitos del metabolismo y no es el único trastorno capaz de producir ambigüedad genital. (16) Es una enfermedad autosómica recesiva en la que los varones y hembras son afectados por igual. (1-4
Desde el punto de vista clínico humoral el diagnóstico del déficit de 21-OH está determinado por el incremento del precursor del cortisol 17-OHP, en presencia de virilización, acompañado o no de pérdida de sal. (16,17
El número de muertes potencialmente evitables con el programa neonatal en pacientes con HSC por deficiencia 21-OH es muy variable, hasta un 10 % según las series, siendo difícil de estimar. En cualquier caso, niños varones con el fenotipo de pérdida salina tienen más probabilidades de sufrir un retraso en el diagnóstico, ya que pueden pasar desapercibidos en la primera exploración física. (12)
A pesar de que nuestra serie exhibe diferencias en cuanto al sexo de los casos diagnosticados con HSC por déficit de 21- OH, cabe resaltar que en Cuba desde el inicio del programa no se ha encontrado una preponderancia en relación al sexo, reportándose una proporción mantenida F:M de 1:1; por lo que se considera que pudiera deberse a la existencia de varones afectados con la forma virilizante simple que pudieran encontrarse aún sin diagnóstico. (5)
No obstante, en los países que no cuentan con un programa de pesquisaje neonatal establecido, se detectan una mayor cantidad de féminas afectadas, lo que hace suponer la existencia de una mortalidad por HSC no reconocida entre los varones.
El diagnóstico molecular en la HSC perfila el asesoramiento genético y permite la aplicación de una conducta preventiva prenatal y postnatal en casos con riesgo de formas severas de la enfermedad. (18) Básicamente consiste en determinar la anomalía en el gen CYP21A2 que codifica la enzima 21-hidroxilasa. (17,19
Al utilizar las posibilidades brindadas por el registro de casos estudiados con HSC que permanece en el laboratorio de biología molecular del Centro Nacional de Genética Médica (CNGM), se pudo determinar que de los casos que resultaron positivos para la mutación estudiada, en su mayoría presentaban al menos una de las mutaciones que actualmente se estudian en el laboratorio del centro (P30L, I2, del 8pb, G318X), lo que los convirtió en individuos genotípicamente heterocigóticos compuestos. A pesar de que en ninguno de los casos se encontró como dato positivo, consanguinidad en la familia, se supone que la frecuencia de heterocigóticos compuestos en la población general es elevada.
Si se analiza la frecuencia de las mutaciones detectadas en la población analizada se comprobará la concordancia con lo reportado por países de la región, como Colombia, donde las mutaciones más frecuentes correspondieron a IVS2-12A/C-G (intrón 2) con 26,7 %, G318X (21,5 %) e I172N que junto a V281L se encontraron en un 12,1 %. (20)
En la población española, Plensa Nebot, reportó un orden de frecuencia de las mutaciones I2, P30L, del 8pb, G318X e I172N muy similar a lo encontrado en la población de este estudio, aunque los valores de frecuencia fueron para cada una de las mutaciones antes mencionadas, superiores a los de este estudio. (15)
La mutación I172N aunque generalmente se relaciona a un fenotipo virilizante simple son varios los reportes en los que se ha visto asociada tanto a formas clásicas como a formas no clásicas, lo cual discrepa de estos resultados. Como se ha demostrado, las características inusuales del locus, así como las mutaciones que se presentan dificultan un análisis real de la relación entre genotipo y fenotipo. Además, la determinación de la ubicación (cis o trans) de las mutaciones en cada uno de los alelos constituye una limitante para ser más precisos en este análisis. (15,21,22
Aunque Diez y cols. describen según su experiencia, que I172N se asocia a la forma virilizante simple, se comprueba una actividad enzimática residual que evita la pérdida salina, pero sí puede existir descompensación electrolítica y una respuesta de renina, por lo que sugieren se sea muy preciso al momento del diagnóstico. (22)
La mutación I172N en heterocigosis compuesta con G318X se presentó con un fenotipo virilizante simple, lo que está en concordancia con lo planteado por la gran mayoría de los investigadores que coinciden en que en los individuos heterocigotos compuestos, por lo general, tienen un fenotipo compatible con la presencia de la más leve de las mutaciones. (18,19,23,24
En esta investigación, no fue posible establecer una adecuada relación genotipo-fenotipo en todos los casos, a pesar de que los resultados obtenidos coinciden en que I172N se presentó en la totalidad de los pacientes con las formas clásicas de la enfermedad, independientemente de su asociación con otras mutaciones. Se considera necesario estudiar a otros familiares, fundamentalmente a los progenitores de los individuos afectados heterocigóticos compuestos, analizar la segregación y a partir del conocimiento de los genotipos de los parentales, deducir la ubicación de las mutaciones en cada uno de los alelos; así como, inferir de acuerdo a los niveles de actividad de la enzima P450c21 reportadas en los estudios realizados in vitro e in vivo, la repercusión fenotípica de las mutaciones cuando se presentan asociadas a otras de mayor o menor gravedad. Además, se deben conocer las mutaciones responsables de las manifestaciones clínicas presentes en los pacientes con HSC por déficit de 21 OH porque orienta en el manejo de individuos afectados, permite reconocer las formas clínicas severas de la enfermedad, y de esta manera se podrá asesorar a familiares y pacientes, con vistas a tomar las medidas preventivas de manera oportuna.
Por lo tanto, en la actualidad, cobra fuerza el análisis molecular como método de gran utilidad para el diagnóstico siempre que sea posible; fundamentalmente en aquellos casos dudosos, así como, los de difícil manejo por la evolución clínica. Favorece, además, poder brindar un adecuado asesoramiento genético, así como una mayor fiabilidad en todo ese complejo proceso.

