










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La enfermedad de Alzheimer (EA) es un trastorno neurodegenerativo caracterizado por la disminución progresiva de la capacidad cognitiva, que influye negativamente en la ejecución de actividades cotidianas. La edad es uno de los principales factores de riesgo de esta enfermedad, donde más del 95% de los pacientes norteamericanos que la padecen son ancianos mayores de 65 años (Chawla y Parikh, 2020). Se espera que su prevalencia incremente de 30 millones de personas en el 2010 a 113 millones en el 2050 (Alzheimer´s Association Report, 2021).
Entre las características fisiopatológicas de la enfermedad se encuentran la acumulación de placas β-amiloides (Aβ) en el exterior de las neuronas, y de nudos neurofibrilares en su interior. Las placas se forman por la acumulación de oligómeros de Aβ, los cuales contribuyen a la neurodegeneración al interferir en la comunicación interneuronal establecida mediante la sinapsis (Kurt, et al, 2001). Los nudos neurofibrilares se forman por la acumulación de inclusiones filamentosas en los cuerpos celulares y las dendritas proximales. En neuronas sanas, la proteína tau se asocia con los microtúbulos, y modula la estabilidad de los ensamblajes de tubulina. En la EA, la proteína tau se hiperfosforila, pierde su asociación con los microtúbulos y forma filamentos helicoidales pareados que forman los nudos neurofibrilares (Grundke-Iqbal y col., 1986).
Entre las principales causas de la EA se encuentran alteraciones producidas en los sistemas de neurotransmisores presentes en el Sistema Nervioso Central (SNC) (Cheng y col., 2021), especialmente en el sistema colinérgico (Hampel y col., 2018). De hecho, la mayoría de los medicamentos disponibles para el tratamiento de la EA están dirigidos a las afectaciones que sufren individuos enfermos en la sinapsis colinérgica del SNC (Tanaka y col., 2021). Aunque estos fármacos poseen efectos positivos de corta duración y no detienen la progresión de la enfermedad, constituyen una alternativa importante en terapias paliativas con enfoque polifarmacológico (Kabir y col., 2020).
La presente revisión bibliográfica tiene como objetivo brindar una panorámica sobre la disfunción colinérgica observada en los pacientes de la EA y que sustenta la hipótesis colinérgica en su fisiopatología. Además, se profundiza en los inhibidores de la actividad colinesterasa de uso clínico e innovadores, como alternativa de terapia sintomática para la EA.
Sinapsis química
El término sinapsis fue introducido por Sherrington (1987), quien la definió como una zona de contacto especializada en la transmisión de la información entre dos neuronas (Foster y Sherrington, 1897). Actualmente, este término también se utiliza para describir las conexiones de las neuronas con las células efectoras y las células receptoras.
Según el criterio morfofuncional, la sinapsis puede clasificarse como eléctrica o química. La sinapsis eléctrica permite la transmisión de señales despolarizantes rápidas y estereotipadas, a diferencia de la sinapsis química, mediante la que se transmite una mayor variabilidad de señalizaciones, y se producen respuestas más complejas. Esta última puede ser mediada por potenciales excitatorios e inhibitorios, y producir cambios en la conductancia y el potencial de la membrana postsináptica, que duran desde pocos milisegundos hasta minutos. Además, en la sinapsis química está presente el fenómeno de amplificación de las señales neuronales; una pequeña terminal presináptica puede alterar la respuesta de una neurona postsináptica de mayores dimensiones. Por estas razones, la mayoría de las sinapsis presentes en el SNC son químicas.
La sinapsis química no posee continuidad estructural; las células pre- y postsináptica están separadas por la hendidura sináptica, que puede alcanzar hasta 40 nm (Kandel y col., 2013). Transmisión del impulso nervioso depende de la difusión de un neurotransmisor a través de la hendidura sináptica.
Transmisión del impulso nervioso
La secuencia de eventos que desencadena la transmisión del impulso nervioso en la sinapsis química, generalmente, ocurre de forma unidireccional. Como evento previo a este fenómeno, se forman las vesículas sinápticas y se llenan de gran cantidad de moléculas neurotransmisoras. Ante la descarga de un potencial de acción en la neurona presináptica, los canales de Ca2+ dependientes de voltaje se abren y permiten el influjo de este ion en la terminal presináptica. Esto incrementa las concentraciones intracelulares de Ca2+, y permite la fusión de las vesículas con la membrana plasmática de las zonas activas de la membrana presináptica.
Las moléculas neurotransmisoras en el interior de las vesículas son liberadas hacia la hendidura sináptica por exocitosis, difunden a través de la misma y se unen a su receptor en la membrana postsináptica. Esta unión desencadena la apertura o cierre de canales iónicos y altera la conductancia y el potencial de membrana. De esta forma, se establecen las condiciones para la descarga de un potencial de acción en la neurona postsináptica. La subsecuente remoción del neurotransmisor mediante degradación enzimática o su secuestro por células gliales, finaliza su acción (Purves y col., 2018).
Sinapsis colinérgica
La Acetilcolina (ACh) es una amina cuaternaria con función neurotransmisora presente en varios tipos de sinapsis de los sistemas nerviosos periférico y central. Su síntesis requiere de la enzima colinoacetiltransferasa (ChAT), y de los precursores acetilCoA y colina, en el citosol de la terminal axónica. La acetilCoA se sintetiza como resultado del metabolismo celular; mientras que la colina es transportada hacia el interior de la terminal axónica desde el fluido extracelular, mediante un transportador específico dependiente de Na+. Una vez concluida la síntesis del neurotransmisor, este es internalizado en vesículas presinápticas mediante un transportador vesicular que intercambia iones H+ por ACh. Es por esta razón que la producción de ACh está limitada por la concentración intracelular de colina, la cual es dependiente del transporte activo de este precursor al interior de la célula (Harilal y col., 2020).
Las neuronas colinérgicas y algunas no colinérgicas, sintetizan acetilcolinesterasa (AChE) (Bear y col., 2007). Esta enzima es secretada en la hendidura sináptica, y se asocia a la membrana de la terminal axónica postsináptica colinérgica. La misma, degrada la ACh en colina y ácido acético rápidamente, debido a su alta eficiencia catalítica (Quinn, 1987), mecanismo esencial que regula la concentración del neurotransmisor en la hendidura sináptica durante la transmisión colinérgica. La colina resultante de esta degradación es secuestrada nuevamente por la terminal axónica y reutilizada en la síntesis de ACh.
Enzima acetilcolinesterasa
La AChE es una enzima que existe en dos formas moleculares: una forma asimétrica monomérica (G1), localizada en la unión neuromuscular; y una forma globular tetramérica (G4), anclada a la membrana de dominios hidrofóbicos. Esta última forma es la que se encuentra con mayor frecuencia en el SNC y su proporción suele modificarse durante el desarrollo de alguna enfermedad neurodegenerativa. Por ejemplo, durante la EA se ha descrito una pérdida selectiva de la forma G4 en algunas regiones corticales y en el hipocampo (Holzgrabe y col., 2007).
Estudios sobre la modelación molecular y mutagénesis en sitios específicos de la AChE, han evidenciado que esta enzima posee tres dominios específicos con residuos de aminoácidos (aa). Estos residuos contribuyen a la selectividad de la enzima por ciertos inhibidores (Radić y col., 1993), y a la especificidad por el sustrato (Vellom y col., 1993). La primera estructura cristalográfica de la AChE, fue obtenida por (Sussman y col., 1991) de Torpedo californica. La misma consiste en 537 aa, y contiene 12 hojas plegadas β rodeadas de 14 hélices α (PDB ID: 1EVE).
La zona más externa de interacción con el sustrato se denomina sitio aniónico periférico (PAS, de sus siglas en inglés), formado por los residuos Trp 84, Trp 279 y Phe 330 (Sussman y col., 1991). Además, su entrada se encuentra alineada con numerosos aa aromáticos que incluyen: Tyr 70, Tyr 72, Tyr 121, Tyr 279 y Tyr 334. Esta zona incrementa la eficiencia catalítica de la enzima al orientar al sustrato hacia el centro activo (Sharma y col., 2020). Adicionalmente, se ha planteado que esta región promueve la agregación del péptido Aβ, por lo que se piensa que los inhibidores específicos por este sitio también inhiben la deposición de placas Aβ (Inestrosa y col., 1996).
La zona más interna del centro activo consiste en cuatro subsitios: el bolsillo de acilación, el subsitio aniónico, la cavidad oxianiónica y el sitio catalítico (CAS). Este último, también conocido como sitio esteárico, es el sitio donde ocurre la hidrólisis de la ACh, y está formado por tres aa en T. californica: Ser 200, Glu 327 e His 440 (Sussman y col., 1991). El proceso de hidrólisis ocurre en dos etapas sucesivas y muy rápidas de acilación y desacilación (Figura 1).
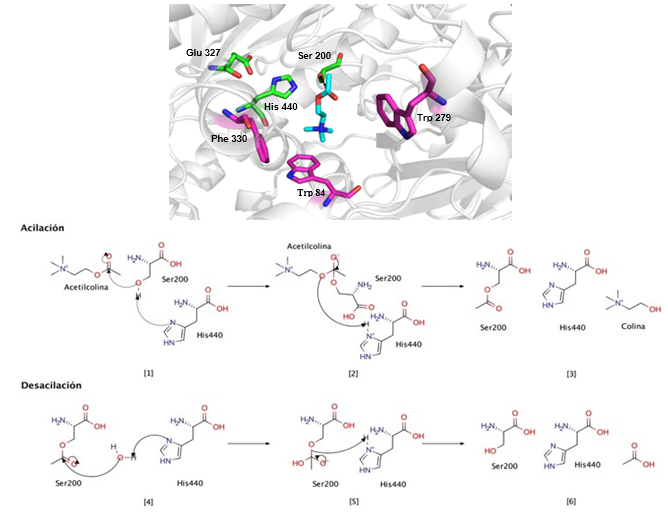
Fig. 1 Mecanismo catalítico de la enzima AChE de Torpedo californica en presencia del sustrato ACh. Se observan las interacciones con los aa pertenecientes al CAS (Ser 200, Glu 327 e His 440), en verde, y al PAS (Trp 84, Trp 279 y Phe 330), en magenta, con el sustrato ACh, en azul. Se muestran los reaccionantes ([1] y [4]), intermediarios ([2] y [5]) y productos ([3] y [6]) de los mecanismos de acilación y desacilación, respectivamente.
La cavidad oxianiónica, formada por los residuos Gly 121, Gly 122 y Ala 204 (Ordentlich y col., 1998), es un factor determinante en la geometría del centro activo de la enzima. Este sitio acomoda el grupo acetil del sustrato ACh hacia el CAS para la hidrólisis, y la estabilización del complejo enzima-sustrato (Zhang y col., 2002). Por otra parte, el bolsillo de acilación de la AChE es responsable de la especificidad por el sustrato ACh. En T. californica, este subsitio consiste en los residuos aromáticos Trp 233, Phe 288, Phe 290 y Phe 331 (Bajda y col., 2013). Adicionalmente, el subsitio aniónico de la AChE es responsable de acomodar la amina cuaternaria de la ACh, y guiar al sustrato hacia el interior del centro activo. En T. californica, este subsitio está conformado por los residuos Trp 84, Glu 199 y Phe 330 (Sharma y col., 2020).
Hipótesis colinérgica
La hipótesis colinérgica se basa en un grupo de características neuropatológicas presentes en la transmisión colinérgica de los pacientes con EA. Entre estas características se encuentran: la disminución de neuronas colinérgicas en la corteza cerebral (Bowen y col., 1976); que el núcleo basal de Meynert en la región basal del cerebro anterior es la fuente de la inervación colinérgica cortical con mayor neurodegeneración en pacientes con EA (Whitehouse y col., 1981); y la demostración de que los antagonistas colinérgicos poseen efectos negativos sobre la memoria y cognición, mientras que los agonistas poseen el efecto contrario (Drachman y Leavitt, 1974).
Adicionalmente, y como parte de la lesión colinérgica, los receptores nicotínicos (nAChR) y muscarínicos de la ACh en corteza, también son afectados (Weinstock, 1995). Por ejemplo, (Nordberg, 2001) planteó que la progresión del déficit cognitivo durante las primeras etapas de la EA, podría estar asociado a la disminución de los niveles de ⍺7-nAChR. Además, cerebros humanos post mortem con EA, han mostrado daños en el acoplamiento de los receptores M1 con la proteína G (Warpman y col., 1993), y una disminución considerable de los receptores muscarínicos ⍺4β2 (Warpman y Nordberg, 1995). Asimismo, (Mulugeta y col., 2003) observaron una reducción significativa de los receptores muscarínicos M4 en el giro dentado y en la región CA4 del hipocampo de pacientes con EA.
La denervación colinérgica actúa de conjunto con otras afectaciones en el desarrollo del déficit cognitivo que se produce en la EA (Figura 2), entre los cuales se incluyen los cambios patológicos mediados por las placas Aβ. Concretamente, se ha descrito que la denervación colinérgica acelera la deposición de Aβ en ratones APP/PS1 y contribuye al desarrollo de la discapacidad cognitiva en la EA (Ramos-Rodríguez y col., 2013). Además, estudios in vitro basados en células sugieren que la incubación de cultivos celulares con la AChE inhibe la actividad 𝛄-secretasa e incrementa los niveles de la Presenilina 1 (PS1, de sus siglas en inglés). (Campanari y col., 2014) Asimismo, la proteína precursora amiloide (APP, de sus siglas en inglés) influye en la expresión de la AChE. (Hicks y col., 2013) observaron que la sobreexpresión de APP695 reprime la expresión de AChE en líneas neuronales humanas. Adicionalmente, estudios in silico han sugerido que los oligómeros Aβ inhiben la actividad de la ChAT y reducen los niveles de ACh en la neurona (Fgaier y col., 2015). Por otra parte, estudios in vitro con proteínas humanas purificadas, describen que los oligómeros Aβ incrementan la actividad hidrolizante de la butirilcolinesterasa (BChE) mediante la formación de los complejos BAβACs, junto a la apolipoproteína E (APOE) (Kumar y col., 2016).
También se ha descrito que la unión del péptido Aβ(1-42) al 𝛂7-nAChR incrementa la actividad sináptica colinérgica, seguido de una dramática inhibición de la misma en el modelo de Drosophila (Hahm y col., 2018), y que ese mismo péptido puede inducir la muerte celular en cultivos de neuroblastoma humano con sobreexpresión de ⍺7-nAChR (Wang y col., 2000). Por su parte, (Fisher, 2012) describió que la estimulación de receptores muscarínicos M1 y M3 promueve el procesamiento no amiloidogénico de APP. Asimismo, (Yi y col., 2020) plantearon que la activación de los receptores metabotrópicos de glutamato-5 mediada por los oligómeros Aβ, regula negativamente la actividad de los receptores muscarínicos M1.
La relación entre el sistema colinérgico central y los cambios patológicos asociados a la proteína tau en la EA, también ha sido investigada (Figura 2). Particularmente, se ha descrito que la denervación colinérgica y la activación de la glucógeno sintasa quinasa 3 (GSK3, de sus siglas en inglés) aceleran la hiperfosforilación de la proteína tau (Zhao y col., 2013). Esto produce el desprendimiento de tau de los mircrotúbulos, lo cual interfiere con los motores de kinesina y perjudica el transporte mitocondrial en el axón neuronal. Por su parte, la activación de los receptores M1 activa la proteína quinasa C (PKC, de sus siglas en inglés) e inhibe la GSK3-β, lo cual produce una reducción significativa de la hiperfosforilación de tau (Agarwal y col., 2011).
La acumulación de tau en el interior de las neuronas tiene efectos destructivos sobre las estructuras sinápticas (Majdi y col., 2020). Se ha descrito que degrada los receptores nicotínicos en la membrana postsináptica mediante la activación de la calpaina-2 en cultivos celulares de hipocampo (Yin y col., 2016). Por otra parte, la estimulación de los receptores ⍺7-nAChR activa la janus quinasa activada 2 (Jak2, de sus siglas en inglés), la cual se asocia con la tirosina quinasa de tipo no receptor Fyn, y permite la activación de la fosfatidilinositol 3-quinasa (PI3K), que fosforila a la porteína quinasa B (Akt, de sus siglas en inglés) y, consecuentemente, inhibe a la GSK3, lo cual disminuye la fosforilación de tau (Bitner y col., 2009).
Como parte de la neurodegeneración observada en la EA, los nudos neurofibrilares intraneuronales de proteína tau hiperfosforilada, pueden ser liberados al ambiente extracelular (Goedert, 1999). Con respecto a esto, (Gómez-Ramos y col., 2006) sugirieron que los receptores muscarínicos interactúan con tau extracelular e incrementan la concentración de Ca2+ intracelular a niveles tóxicos en cultivos celulares de neuroblastoma.
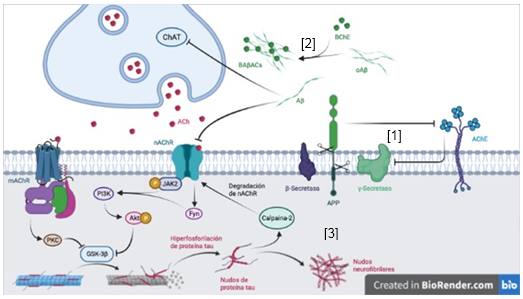
Fig. 2 Relación entre el sistema colinérgico y los cambios patológicos producidos por Aβ y la proteína tau en la EA. [1] La sobreexpresión de APP disminuye la expresión de AChE, en tanto la AChE inhibe la actividad 𝛄-secretasa e incrementa los niveles de PS1. [2] Los oligómeros Aβ forman los complejos BAβACs al interactuar con la BChE. Estos oligómeros inhiben la actividad de la enzima ChAT y de los receptores nicotínicos ⍺7-nAChR. [3] La acumulación de proteína tau hiperfosforilada intracelular activa a la calpaina-2, la cual degrada los receptores nicotínicos de la ACh. La hiperfosforilación de tau puede disminuir mediante la inhibición de la GSK3. A través de la activación de la PKC por los receptores muscarínicos de la ACh; o la cascada de fosforilación que involucra a la JAK2, la PI3K y la Akt. Hecho en BioRender.com
Inhibidores terapéuticos de la actividad colinesterasa
Los inhibidores de la actividad colinesterasa constituyen un amplio grupo de compuestos químicos con características físico-químicas diferentes. Estos inhiben a las enzimas responsables del hidrólisis de la ACh en la hendidura sináptica, e incrementan la disponibilidad del neurotransmisor en el SNC de los pacientes con EA. Por lo tanto, mejoran la neurotransmisión colinérgica central y mitigan el deterioro cognitivo significativamente en los primeros años de tratamiento (Xu, et al., 2021). La interrupción temporal del tratamiento con estos fármacos produce un rápido declive cognitivo y está asociado a un mayor riesgo de ingreso hospitalario (Howard y col., 2015).
La mayoría de las terapias farmacológicas disponibles para enfrentar la EA son principalmente inhibidores de la AChE. (Weiner y col., 2009) Clasificaron los inhibidores reversibles de la AChE en tres categorías: [1] inhibidores dirigidos al centro activo, que interactúan con el subsitio aniónico catalítico en el fondo del mismo; [2] inhibidores de sitios aniónicos periféricos, que interactúan con grupos en la entrada del centro activo; e [3] inhibidores que interactúan con ambos sitios y forman puentes entre ellos.
Inhibidores de las colinesterasas con uso clínico aprobado
A pesar de la creciente incidencia y prevalencia de la EA, sólo existen seis tratamientos aprobados para su uso en el enfrentamiento terapéutico de esta enfermedad (Lalli y col., 2021), de los cuales cuatro son inhibidores de la actividad colinesterasa (Figura 3).
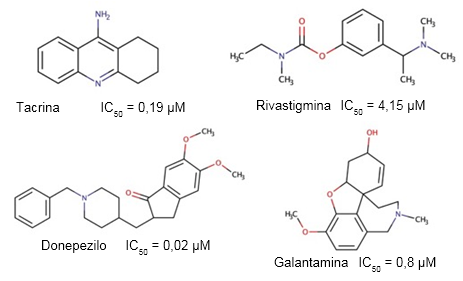
Fig. 3 Estructuras de los inhibidores de la actividad colinesterasa aprobados por la FDA para el tratamiento de la EA, y los valores de IC50 descritos para su actividad inhibitoria sobre la AChE humana (Luo y col., 2005).
Tacrina
La tacrina es una aminoacridina capaz de inhibir tanto la actividad colinesterasa (Ahmed y cols., 2006) como de la histamina N-metil-transferasa. Además, es un antagonista de los receptores muscarínicos M1 y M2, y es capaz de interactuar débilmente con los receptores nicotínicos de la ACh (Freeman y Dawson, 1991). Este fue el primer medicamento inhibidor de la actividad colinesterasa aprobado por la FDA para combatir la EA. Sin embargo, salió rápidamente del mercado debido a la alta incidencia de efectos secundarios como la hepatotoxicidad, y afectaciones gastrointestinales como náuseas y vómitos (Watkins y col., 1994).
A pesar de poseer un uso descontinuado a nivel clínico, la tacrina continúa siendo de interés investigativo para muchos grupos dedicados a la formulación y síntesis de agentes multifuncionales para el tratamiento de la EA. Esto se debe a que su hepatotoxicidad puede ser abolida mediante su unión a otros farmacóforos (Panek y col., 2017). La tacrina constituye por tanto una referencia para el diseño de nuevos fármacos sin efectos hepatotóxicos, y capaces de modular diversas dianas biológicas relacionadas con el progreso y evolución de la EA (Bautista-Aguilera y col., 2020).
Rivastigmina
La rivastigmina es un derivado de fenilcarbamato, capaz de inhibir de manera pseudoirreversible la actividad colinesterasa, con efectos de mayor duración de la que puede cuantificarse en el plasma. Una vez absorbida, su comportamiento es similar al de la ACh y se une tanto al centro activo como al sitio aniónico periférico de la AChE. En lugar de disociarse inmediatamente tras su hidrólisis, la rivastigmina deja el sitio esterático de la AChE carbamilado temporalmente, lo cual inhibe la actividad enzimática (Nguyen y col., 2021).
La rivastigmina es capaz de inhibir ambas colinesterasas, lo cual posee relevancia clínica para el tratamiento de la EA. Esto es debido a que se han descrito elevados niveles de BChE en el hipocampo y la corteza temporal de pacientes con EA, mientras que la actividad de la AChE disminuye en dichas regiones (Perry y col., 1978). Además, la inhibición de ambas colinesterasas incrementa más los niveles de ACh en el cerebro, en comparación con la simple inhibición de una de estas enzimas (Lane y col., 2016). Adicionalmente, es el único de los inhibidores de la actividad colinesterasa empleado para la terapia sintomática de la EA que disminuye sustancialmente la actividad AChE en el líquido cefaloraquídeo (Marco-Contelles y col., 2006).
De hecho, en pacientes con niveles de EA leves a moderados, mejora las funciones cognitivas y su desenvolvimiento en actividades cotidianas. La administración oral de rivastigmina está asociada a efectos tróficos adversos como náuseas, vómitos, dispepsia, astenia, anorexia y pérdida de peso (Müller, 2007). Estos efectos secundarios suelen desaparecer a medida que aumenta el tiempo de consumo del fármaco, pero constituyen la causa principal por la cual los pacientes suelen abandonar el tratamiento.
La rivastigmina puede administrarse mediante parches transdérmicos de manera controlada y continua (Lefèvre y col., 2009, 2007), lo cual genera mayor tolerabilidad y reducción de los efectos secundarios. Esta forma de administración resulta ventajosa, ya que muchos pacientes con EA sufren problemas de deglución, lo cual afecta la administración de fármacos orales a intervalos regulares (Khoury y col., 2018).
Galantamina
La galantamina es un alacaloide terciario de isoquinolina que ha sido obtenido de varias plantas que incluyen algunas especies del género Galanthus, y otros géneros relacionados, que incluyen Narcissus, Leucojun y Lycoris (Shi y Seltzer, 2011). El costo de extracción de esta sustancia es elevado, y las fuentes naturales de las que proviene escasas, por lo que se han desarrollado métodos para su síntesis (Marco-Contelles y col., 2006).
La galantamina es un inhibidor competitivo y reversible, con alta selectividad por la AChE (Scott y Goa, 2000). Aunque el mecanismo de acción de este inhibidor aún no ha sido completamente dilucidado, se ha asociado su efecto terapéutico al incremento de la función colinérgica. Este es el único fármaco comercialmente disponible que, además, modula alostéricamente la subunidad ⍺ de los receptores nAChR y los activa (Woodruff-Pak y col., 2002, 2001). Los nAChR se localizan en terminales de neuronas colinérgicas y no colinérgicas. La activación de estos receptores estimula la liberación de ACh y de otros neurotransmisores como la dopamina, la noradrenalina, el 5-HT y el glutamato. Por eso la acción moduladora alostérica de la galantamina en los nAChR se asocia al incremento de la liberación de estos neurotransmisores en el SNC (Ago y col., 2011).
Este fármaco mejora los síntomas conductuales, el desenvolvimiento en actividades cotidianas y el procesamiento cognitivo con buena eficacia y tolerabilidad. Entre los principales efectos adversos se encuentran náuseas, vómitos, diarrea, anorexia y pérdida de peso (Epperly y col., 2017). Entre los efectos adversos no tróficos que producen la discontinuidad del tratamiento se encuentran mareos y síncopes; aunque estos han sido descritos en la minoría de los pacientes. La mayoría de estas afectaciones ocurren durante el período de escalada de la dosis. El consumo de galantamina junto a los alimentos y la ingesta adecuada de líquido promueve la reducción de estos efectos no deseados (Shi y Seltzer, 2011).
La galantamina se desarrolló inicialmente como un comprimido de liberación inmediata y posteriormente se reformuló como una cápsula de liberación prolongada (Hing y col., 2005). Sin embargo, se han desarrollado varios métodos para mejorar la administración de este fármaco. Por ejemplo, (Wahba y col., 2016) adjuntaron la galantamina a nanopartículas de hidroxiapatita que contenían cerio para la administración selectiva del fármaco a las regiones afectadas del cerebro. Otro ejemplo de optimización de la administración de la galantamina se evidencia en el profármaco Memogain. Este se aplica nasalmente, lo cual permite la llegada más rápida del fármaco al cerebro de pacientes con (Bhattachara y col., 2015) EA. También se han desarrollado parches transdérmicos de galantamina, los cuales poseen un pH moderado, alto contenido del fármaco y un patrón de liberación controlada del mismo, con alto potencial para el tratamiento de la EA (Woo y col., 2015).
Donepezilo
El donepezilo es un derivado de la indanonebenzilpiperidina que actúa como inhibidor reversible y no competitivo de la actividad colinesterasa en el SNC y otros tejidos (Brewster II y col., 2018). Posee una mayor selectividad por la AChE, en comparación con la BChE (Dooley y Lamb, 2000) y es considerado como el fármaco líder para el tratamiento de la EA (Breijyeh y Karaman, 2020). La unión del donepezilo a la AChE inhibe la hidrólisis de la ACh y aumenta la disponibilidad de este neurotransmisor en la hendidura sináptica.
Debido al amplio uso que posee este fármaco en el tratamiento de la EA, los efectos adversos que produce su administración y consumo han sido de gran interés investigativo. En un estudio comparativo de (Hansen y col., 2008), se obervó que la incidencia de los efectos adversos comunes entre los inhibidores de la actividad colinesterasa disponibles para el tratamiento de la EA, son menos frecuentes en el donepezilo y más frecuentes en la rivastigmina. Estos efectos adversos pueden clasificarse en dos categorías: [1] efectos secundarios comunes, observados generalmente en pacientes con EA; y [2] efectos secundarios poco frecuentes o extraordinarios, observados sólo en condiciones especiales o en pequeños grupos de pacientes bajo tratamiento con otros medicamentos concomitantes y que sufren de otras patologías. Las reacciones adversas más frecuentes producidas por el tratamiento con donepezilo incluyen: problemas cardiovasculares (Kuwahata y col., 2021); alteraciones del sistema digestivo, hemáticas y linfáticas; cambios metabólicos y nutricionales; problemas musculares y esqueléticos; complicaciones en el sistema respiratorio, piel y apéndices; problemas urogenitales y del SNC. Estos últimos incluyen: agitación, insomnio, confusión, depresión, ansiedad, mareos, vértigo, dolor de cabeza, inquietud y alucinaciones (Cacabelos, 2007) .
(Dubois y col., 2014) observaron una reducción del 45% de la tasa de atrofia del hipocampo tras un tratamiento con donepezilo en pacientes con sospecha de EA prodrómica. Resultados similares fueron obtenidos por, (Cavedo y col., 2017) quienes observaron que la administración de donepezilo en la fase prodrómica de la EA redujo sustancialmente la tasa de atrofia de los núcleos del sistema colinérgico en la región basal del cerebro anteior. Además, se ha descrito que el donepezilo posee un efecto neuroprotector contra las lesión celular neuronal inducida por los oligómeros Aβ (Kimura, Akasofu y col., 2005). Estos resultados sugieren que el donepezilo disminuye el deterioro cognitivo no sólo mediante la mejora de la función colinérgica, sino también mediante la inhibición de la neurodegeneración progresiva en la EA.
Terapias combinadas de inhibidores en uso clínico
La combinación de terapias farmacológicas aprobadas por la FDA constituye una alternativa para mejorar la eficacia del tratamiento de la EA. De hecho, (Matsunaga y col., 2015) sugirieron la combinación de los inhibidores colinesterásicos y la memantina, inhibidor de los receptores NMDA, como un tratamiento beneficioso para pacientes con EA de moderada a grave. También se ha descrito que la combinación de galantamina y memantina, constituye una mejor alternativa en comparación con la monoterapia con galantamina para mejorar la cognición de pacientes con deterioro cognitivo leve debido a la EA (Koola, 2020). Además, se ha observado que la combinación de estos fármacos es superior a la de donepezilo y memantina, en cuanto al mejoramiento cognitivo de pacientes con EA (Koola y col., 2018).
Inhibidores derivados de fuentes naturales en fase de estudio preclínico
Huperzinas
Las huperzinas (Hup) son alcaloides de licopodio, que pueden ser extraídos y aislados de la planta Huperzia serrata (Liu y col., 1986). Esta planta fue utilizada en la medicina natural y tradicional china para aliviar la deficiencia de memoria. Estos compuestos poseen potencialidades para su uso en el tratamiento sintomático de la EA, pues incrementan la disponibilidad de ACh en la hendidura sináptica (Sharma,2019).
Existen dos tipos de Hup: la HupA y la HupB (Figura 4). La Hup A es un fuerte inhibidor reversible de la AChE (Ki = 20 - 40 nM) (Rajendran y col., 2002) que interactúa con los residuos aromáticos presentes en el centro activo de la enzima AChE (Upadhyay y col., 2019). Adicionalmente, la Hup A posee una alta afinidad por la enzima AChE, lo que permite una rápida formación del complejo AChE-HupA, el cual se disocia lentamente (Sharma y col., 2020). En comparación con otros inhibidores de la AChE disponibles, como la rivastigmina, el donepezilo y la tacrina, produce una inhibición más sostenida de la enzima, posee mayor biodisponibilidad oral y es mucho mejor transportada a través de la barrera hematoencefálica (BHE) (Cheng y Tang, 1998).
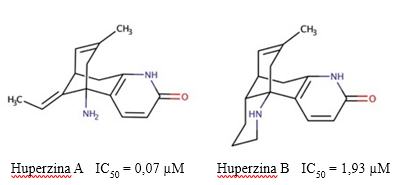
Fig. 4 Estructura química de las huperzinas A y B, y los valores de IC50 descritos para su actividad inhibitoria sobre la AChE (Szypula y col., 2020).
No existen muchos estudios farmacológicos sobre las propiedades de la Hup B, aunque se le conoce por ser un inhibidor reversible con alta selectividad por la AChE (Liu, et al, 1999). Como inhibidor de la AChE, la HupB (IC50 = 1930 nM) es más débil que la HupA (IC50 = 72,4 nM) (Szypula y col., 2020), pero posee un mayor índice terapéutico (Hanin y col., 1991). Adicionalmente, es un agente terapéutico más potente que la galantamina, pero más débil que la fisostigmina (Yan y col., 1987).
Fisostigmina
La fisostigmina, también conocida como eserina, es un alcaloide (Figura 5) aislado de las semillas de Physostigma venenosum, y constituye un prototipo de inhibidor pseudoirreversible de la AChE (IC50 = 1,0 nM) (Zhang y col., 2010), con alta selectividad por esta enzima (Adeniyi y Conradie, 2019). A pesar que este fármaco puede atravesar la BHE, carece de índice terapéutico debido a que es altamente tóxico (Dembitsky y col., 2020), su tiempo de vida medio es muy corto, y posee numerosos efectos adversos. Entre estos últimos se encuentran: diarrea, dolor estomacal, incremento de la salivación y sudoración excesiva (Arens y Kearney, 2019). Estas desventajas produjeron que no fuera aprobada para su uso en el tratamiento de la EA (Coelho y Birks, 2001).

Fig. 5 Estructura química de la fisostigmina y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Zhan y col., 2010).
A pesar de sus efectos adversos, la administración de fisostigmina produce otros efectos positivos, además de incrementar la disponibilidad de ACh en la sinapsis colinérgica. Por ejemplo, (Bitzinger y col., 2019) observaron que la administración de fisostigmina en ratas Wistar disminuyó significativamente el estrés oxidativo al inhibir la producción de especies reactivas del oxígeno (ROS, de sus siglas en inglés). Estas propiedades de la fisostigmina resultan de gran interés clínico y potencian la continuidad de las investigaciones relacionadas con este compuesto.
Sinapina
La sinapina es un éster de ácido sinápico y colina (Figura 6) que puede extraerse de las semillas de plantas crucíferas como Raphanus sativus. Este compuesto se encuentra estructuralmente muy relacionado con la ACh, y actúa como un inhibidor competitivo de la enzima AChE (Hasan y col., 1981).
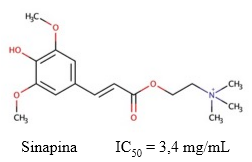
Fig. 6 Estructura química de la sinapina y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Ferreres y col., 2009).
La fuerte capacidad inhibitoria de la sinapina sobre la AChE en modelos in silico e in vitro (Yates y col., 2018), y ex vivo; aunque esta inhibición fue más débil en ensayos realizados en plasma (He y col., 2008). Adicionalmente, (Ferreres y col., 2009) observaron valores de IC50 = 3,399 mg/mL en ensayos enzimáticos con extractos secos liofilizados de Brassica oleraceae que contenían 22,8 µg/mL de sinapina.
Galangina
La galangina es un flavonol (Figura 7) derivado de los rizomas de la planta Alpiniae officinarum, que ha mostrado potente actividad inhibitoria de la AChE (IC50 = 120 µM) (Guo y col., 2010). La naturaleza flavonoide de este compuesto le confieren una superficie de polaridad topológica muy baja y un coeficiente de partición agua-lípido muy alto. Estas características permiten que la galangina atraviese la BHE con relativa facilidad y actividad potencial. (Baker, 2022)
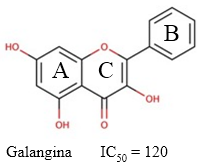
Fig. 7 Estructura química de la galangina y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Guo y col., 2010).
La actividad inhibitoria de la galangina sobre las enzimas colinesterasas está altamente relacionada con sus características estructurales. De hecho, (Xie y col., 2014) observaron que la hidroxilación del anillo A de los flavonoides incrementa el potencial inhibitorio de estos compuestos. Esto probablemente se deba a la formación de puentes de hidrógeno entre los grupos hidroxilo en ese anillo aromático y el centro activo de la enzima.
La actividad anticolinesterásica de la galangina ha sido evaluada en modelos in vivo. Por ejemplo, (Kilic y col., 2019) observaron que la galangina disminuyó el deterioro de la memoria inducido por la administración de escopolamina y mecamilamina en ratas Sprague Dawley. Estos autores sugirieron que la galangina producía dicho efecto debido a que no sólo inhibía la actividad colinesterásica, sino que interactuaba con los receptores nicotínicos y muscarínicos de la acetilcolina. Además, observaron que la administración aguda de galangina aumentó las concentraciones de ACh en el hipocampo de los individuos tratados y no tratados con escopolamina o mecamilamina.
Adicionalmente, (Katalinić y col., 2010)observaron que la galangina se une a la BChE con una preferencia 12 veces mayor que a la AChE. Estos autores comentaban que dicha afinidad podía deberse a que el anillo aromático B de este flavonoide no se encuentra hidroxilado. Sus resultados permiten sugerir que la galangina es un compuesto prometedor y relevante en la búsqueda de inhibidores de la BChE. A pesar de las potencialidades que posee la galangina como inhibidor de las enzimas colinesterasas, no se han realizado investigaciones sobre su toxicidad en ensayos preclínicos o clínicos (Sharma, 2019).
Cardanol
El cardanol es un lípido fenólico no isoprenoide (Figura 8) obtenido del extracto de la cáscara de Anacardium occidentale, que mostró cierto grado de inhibición de la AChE (de Paula y col., 2009). No se han realizado muchos estudios farmacológicos para evaluar sus propiedades anticolinesterásicas, aunque se ha descrito que posee un valor de IC50 = 4,0 µM (Stasiuk y col., 2014). Adicionalmente, su toxicidad no ha sido investigada en ensayos preclínicos ni clínicos (Sharma, 2019).
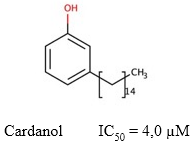
Fig. 8 Estructura química de la cardanol y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Uliassi y col., 2021).
Inhibidores sintéticos en fase de estudio preclínico
Fenserina
La fenserina es un fenilcarbamato derivado de la fisostigmina (Figura 9), con actividad inhibitoria selectiva por la AChE (Al-Jafari y cols., 1998). Sobre este compuesto, (Tabrez y Damanhouri, 2019) estimaron que posee una IC50 = 13,93 nM, y plantearon que su mecanismo de inhibición sobre la enzima es mixto. Este compuesto se une tanto a la enzima libre como al complejo enzima-sustrato, y con valores de Ki de 0,39 µM y 0,21 µM, respectivamente.
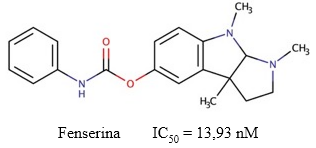
Fig. 9 Estructura química de la fenserina y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Tabrez y Damanhouri, 2019).
Este fármaco también es capaz de reducir la producción de APP en modelos in vivo e in vitro (Ahmad y cols., 2016), además de mejorar la memoria y el aprendizaje de perros y ratas. Adicionalmente, las pruebas realizadas para su uso en el tratamiento de la EA rindieron resultados exitosos en ensayos clínicos de fase II (Sharma, 2019). Su toxicidad es menor que la reportada para otros inhibidores terapéuticos de la actividad colinesterasa como la tacrina y la fisostigmina (Greig y col., 2000).
Tolserina
La tolserina es un fenilcarbamato derivado de la fisostigmina, cuya estructura difiere de la fenserina por la presencia de un grupo metilo en la posición de su fracción fenilcarbamoil (Yu y col., 1999) (Figura 10). Posee potencialidades para su uso en el tratamiento de la EA, ya que es fácilmente absorbido por las membranas y puede ser aplicado tópicamente en la conjuntiva. Además, es capaz de traspasar la BHE y es utilizada para el tratamiento de toxicidad anticolinérgica severa (Ahmad y cols., 2016). Su potencia y selectividad en la inhibición de la AChE humana es mayor (IC50 = 10,3 nM) en comparación con las descritas para la fenserina y la fisostigmina (Kamal y col., 2000).
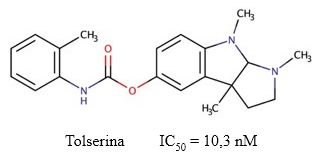
Fig. 10 Estructura química de la tolserina y el valor de IC50 descrito para su actividad inhibitoria sobre la AChE (Yu y col., 2011).
Eserolina
La eserolina es un metabolito de la fisostigmina (Figura 11) que actúa como agonista opioide. A diferencia de la fisostigmina, su efecto sobre la inhibición de la AChE es limitado y reversible (Galli y col., 1982), con mayor afinidad por esta enzima que por la BChE (Motel y col., 2013) (Fürst y col., 1982). Adicionalmente, Galli y col. (1996) obtuvieron un valor de IC50 = 1,0 µM para este compuesto. El desarrollo de este fármaco fue interrumpido debido a su neurotoxicidad, pues produce la muerte de células neuronales mediante un mecanismo que involucra la pérdida de ATP (Somani y col., 1990).
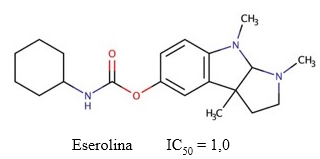
Fig. 11 Estructura química de la eserolina. Se muestra el valor de IC50 descrito para la actividad inhibitoria de este compuesto sobre la AChE (Galli y col., 1996).
Cymserina
La cymserina (Figura 12) es un inhibidor reversible de la BChE (IC50 = 51 nM) (Yu y col., 2011), que permite incrementar los niveles de ACh en el cerebro sin producir efectos adversos como temblor, lagrimeo, salivación o afectaciones tróficas (Greig y col., 2002). Este compuesto posee un mecanismo de inhibición no competitivo y mixto sobre la BChE (Kamal y col., 2006), lo cual probablemente sea consecuencia de las múltiples interacciones hidrofóbicas secundarias y los puentes de hidrógenos que forma con el sitio activo de la enzima (Luo y col., 2005). Adicionalmente, la cymserina y algunos de sus derivados y análogos reducen los niveles de APP y Aβ (Kamal y col., 2008), y poseen propiedades antiinflamatorias (Reale y col., 2014).
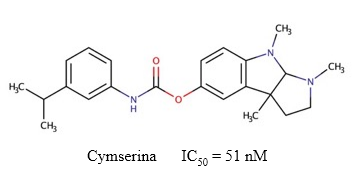
Fig. 12 Estructura química de la cymserina y el valor de IC50 descrito para su actividad inhibitoria sobre la BChE (Yu y col., 2011).
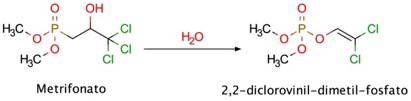
Metrifonato
El metrifonato es un inhibidor organofosforado de la AChE con acción prolongada, y se utiliza en el tratamiento de la esquistosomiasis (Nordgren y col., 1981). Este fármaco puede mejorar la neurotransmisión colinérgica mediante un metabolito farmacológicamente activo, el 2,2-diclorovinil-dimetil-fosfato, obtenido por una hidrólisis no enzimática del metrifonato (Figura 13). La administración de metrifonato una vez al día mejora la función cognitiva de pacientes con EA de media a moderada (Cummings y col., 1998). La tolerabilidad de este fármaco es buena, pero su uso produce efectos secundarios adversos a largo plazo, entre los que se incluyen problemas con la transmisión neuromuscular y parálisis respiratoria (López-Arrieta y Shneider, 2010). Por esta razón, el desarrollo de este fármaco fue interrumpido durante los ensayos clínicos de fase III.
Inhibidores híbridos
Debido a que la EA posee carácter multifactorial, es necesario el diseño de pequeñas moléculas capaces de interactuar con múltiples dianas terapéuticas implicadas en la patología de esta enfermedad. Entre las propiedades ideales que deben presentar estas nuevas entidades se encuentran: [1] un elevado índice terapéutico; [2] la capacidad de atravesar la BHE; [3] tener características neuroprotectoras y antioxidantes, así como propiedades similares a las de los fármacos con uso clínico aprobado (Bautista-Aguilera y col., 2020). Es por esta razón que es necesario el desarrollo de nuevos compuestos para terapia multiblanco, a partir de farmacóforos promisorios conocidos; especialmente aquellos cuyo uso pueda verse limitado por sus efectos adversos.
Donepezilo-AP2238
AP2238 es un derivado de cumarina (Figura 14) considerado como el primer fármaco desarrollado con la capacidad de interactuar con los dos sitios aniónicos de la AChE humana (Piazzi y col., 2003). Se ha descrito que este compuesto y algunos de sus análogos poseen un grado de actividad similar al donepezilo en cuanto a la inhibición de la AChE (Piazzi y col., 2007), y mayor en cuanto a la inhibición de la toxicidad mediada por los oligómeros Aβ (Tarozzi y col., 2014).
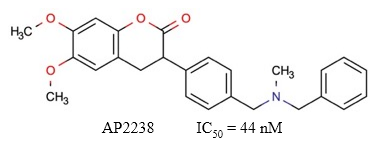
Fig. 14 Estructura química de la cymserina. Se muestra el valor de IC50 descrito para la actividad inhibitoria de este compuesto sobre la AChE (Rizzo y col., 2010).
Rizzo y col. (2010) investigaron sobre las características y la actividad de 22 compuestos híbridos de donepezilo y AP2238 (Figura 15). El diseño de estos compuestos perseguía obtener nuevos inhibidores de la AChE, que a su vez inhibiesen la agregación de los oligómeros Aβ. En su estudio, las dos moléculas con mayor actividad poseían una cadena alquílica de cinco átomos de carbono, y un grupo amino presente en el final de la cadena. Esta estructura permitía una mayor interacción con los sitios aniónicos periféricos de la AChE, lo cual mejoró significativamente la inhibición de la agregación de los oligómeros Aβ, con respecto a los compuestos de referencia.

Fig. 15 Estructura química de dos híbridos promisorios de Donepezilo y AP2238 (Rizzo y col., 2010). Se muestran los valores de IC50 para su actividad inhibitoria sobre la AChE, y los grupos aportados por los precursores en su diseño. Compuesto 1: (2E)‐2‐[(4‐{[bencil(metil)amino]metil}fenil)metilideno]‐6‐{[5‐(dietilamino)pentil]oxi}‐1,2,3,4‐tetrahidronaftalen‐1‐ona; Compuesto2:(2E)‐2‐[(4‐{[bencil(metil)amino]metil}fenil)metilideno]‐6‐{[5‐(piperidin‐1‐il)pentil]oxi}‐1,2,3,4‐tetrahidronaftalen‐1‐ona.
Donepezilo-Tacrina
A pesar de sus fuertes efectos secundarios, la estructura de la tacrina ha sido ampliamente utilizada en la química medicinal para el diseño de híbridos o compuestos multiblanco sin efectos tóxicos (Oset-Gasque y Marco-Contelles, 2020). Eso se debe a que los efectos secundarios producidos por la administración de tacrina pueden ser disminuidos mediante su unión a otros farmacóforos (Spilovska y col., 2017).
Entre los farmacóforos utilizados para el diseño de nuevos compuestos híbridos junto a la tacrina se encuentra el donepezilo (Figura 16A). Por ejemplo, (Camps y col., 2008) diseñaron una serie de híbridos de tacrina y donepezilo que interactúan simultáneamente con los sitios activos periféricos y mediales de la AChE. Estos híbridos no solo inhiben la actividad de la AChE (IC50 = 0,27 nM)y la BChE (IC50 = 66,3 nM), sino también la deposición Aβ. Estos compuestos son sintetizados por la combinación de la 6-clorotacrina con la fracción indanona del donepezilo, e inhiben a la AChE con mayor eficiencia que sus compuestos parentales.

Fig. 16 Estructura química de algunos compuestos híbridos promisorios de Donepezilo y Tacrina con capacidad inhibitoria sobre la actividad colinesterasa. Se muestran los valores de IC50 descritos para cada compuesto sobre la AChE, y los grupos aportados por los precursores en su diseño. A: 2‐[(1‐{3‐[(6‐cloro‐1,2,3,4‐tetrahidroacridin‐9‐il)amino]propil}piperidin‐4‐il)metil]‐5,6‐dimetoxi‐2,3‐dihidro‐1H‐indeno‐1‐ona (Camps y col., 2008) ; B: 2‐{9‐[(acridina‐9‐il)amino]nonil}‐2,3‐dihidro‐1H‐isoindol‐1,3‐diona (Alonso y cols., 2005) C: N‐[2‐(1‐bencilpiperidin‐4‐il)etil]‐2‐[(1,2,3,4‐ tetrahidroacridina‐9‐il)amino]acetamida (Shao y col., 2004).
Adicionalmente, (Alonso y cols., 2005) diseñaron y sintetizaron un compuesto inhibidor potente y selectivo de la AChE (IC50 = 2,4 nM), capaz de desplazar al propidio (inhibidor reversible del PAS) en un ensayo de competencia (Figura 15B). Estos autores sugirieron que la fracción ftilamida presente en este compuesto actuaba como un ligando eficiente del sitio aniónico periférico de la enzima, lo cual permitía una mejor interacción. Además, este híbrido resultó ser mucho más eficiente que sus precursores.
Otro ejemplo de compuestos híbridos de donepezilo y tacrina con potencial inhibitorio de la actividad colinesterasa es el obtenido por (Shao y col.,2004). Estos autores lograron sintetizar un inhibidor híbrido de la AChE mucho más potente y selectivo que la tacrina, con una IC50 = 6,0 nM. El estudio in silico realizado por estos autores sugirió que este ligando interactuaba con el centro activo de la enzima mediante la formación de puente de hidrógeno.
Huprinas
Las huprinas son compuestos híbridos que combinan la estructura carbocíclica de la HupA con la subestructura 4-aminoquinolina de la Tacrina (Camps y col., 2000). La racionalidad de desarrollar esta familia de compuestos híbridos consiste en generar posibles fármacos con mayor selectividad y actividad anticolinesterásica, e incrementar el índice terapéutico de la tacrina. Existen cuatro tipos de huprinas: la huprina X, la huprina Y, la huprina W y la huprina Z (Figura 17).
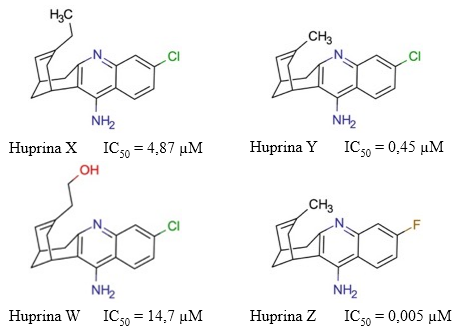
Fig. 17 Estructura química de las huprinas X, Y, W y Z. Se muestran los valores de IC50 descritos para la actividad inhibitoria de estos compuestos sobre la AChE (Ros y col., 2001), (Alcalá y col., 2003), (Son y col., 2019).
La huprina X es un inhibidor reversible de la AChE (IC50 = 4,87 µM) con mayor afinidad que sus compuestos parentales y el Donepezilo (Giménez-Llort y col., 2017). Además, es capaz de incrementar los niveles de ACh con mayor eficiencia que la Tacrina (Martínez y Castro, 2006). Adicionalmente, este compuesto muestra efectos agonistas sobre los receptores nicotínicos y muscarínicos M1 de la ACh (Roman y col., 2002, 2004). Con respecto a esto, (Roman y col., 2005) comentaron que este efecto agonista de los receptores nicotínicos de la ACh parece ser principalmente por la capacidad de este compuesto de inhibir la actividad colinesterasa. Asimismo, en ese mismo artículo plantearon que la huprina X poseía mayor potencial inhibitorio sobre la AChE que la galantamina.
La huprina Y es un inhibidor reversible de la AChE más potente (IC50 = 0,45 µM) que la huprina X (Ros y col., 2001). Este compuesto también ha mostrado efecto neuroprotector en diferentes modelos in vitro e in vivo, de daño por excitotoxicidad (Canudas y col., 2003). En contraste, (Jordá y col., 2004) observaron que la administración de huprina Y no revirtió la pérdida de viabilidad neuronal ni previno el incremento de las características apoptóticas inducidas por colchicina. Es por esta razón que dichos autores sugieren que el fármaco no previene la neurodegeneración inducida por las alteraciones del citoesqueleto.
No existen muchos estudios farmacológicos sobre las propiedades de la huprina W y la huprina Z. Sobre la huprina W, (Nachon y col., 2013) observaron que la huprina W y la tacrina interactuaban en posiciones muy similares en el centro activo de las enzimas AChE y BChE humana. Asimismo, comentaban que esta peculiaridad podría deberse a la conservación de las interacciones clave entre las enzimas colinesterasas y dichos inhibidores. Sin embargo, también se percataron que la huprina W formaba más interacciones con el centro activo de la AChE humana, en comparación con la tacrina. Este resultado sugiere que la huprina W posee una mayor afinidad por la enzima que su precursor. Adicionalmente, Son y col. (2019) estimaron que este compuesto poseía un valor de IC50 = 14,7 µM.
Por otro lado, la huprina Z es un inhibidor reversible de la enzima con mayor potencial inhibitorio (IC50 = 5,09 nM) sobre la AChE humana que sus precursores tacrina y HupA (Alcalá y col. 2005). Adicionalmente, (Alcalá y col. 2005) observaron que tanto la huprina Y como la huprina Z estimulaban significativamente la acumulación de fosfatos de inositol de manera dependiente a la concentración. Asimismo, plantearon que la reversión de dichos efectos mediante diferentes antagonistas sugería que estos compuestos activaban a los receptores muscarínicos. Sin embargo, advertían que otros mecanismos celulares podrían estar implicados.
CONCLUSIONES
Los inhibidores de la actividad colinesterasa constituyen terapias farmacológicas paliativas que permiten disminuir el deterioro cognitivo generado por la EA. Sin embargo, la mayoría de estos compuestos producen efectos secundarios severos que han limitado su éxito terapéutico para enfrentar dicha enfermedad neurodegenerativa. Es por ello que se hace necesaria la investigación y desarrollo de compuestos novedosos con mayor índice terapéutico que permitan enfrentar exitosamente las características neuropatológicas presentes en el sistema colinérgico de individuos con EA.

