










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La conjuntivitis hemorrágica aguda figura entre las enfermedades emergentes y reemergentes causadas por los enterovirus con elevado índice de morbilidad y costo económico a nivel mundial.1,2 La enfermedad es altamente contagiosa, con una tasa de ataque estimada entre el 70 % y 100 % en los miembros de una familia y el 45 %-58 % en la población. Las epidemias generalmente llevan a una congestión significativa de los servicios de atención médica y parálisis de algunos servicios públicos donde confluye un gran número de personas, como las escuelas, centros de trabajo, centros recreativos, productivos, entre otros. En ausencia de las terapias específicas, el único modo efectivo de control consiste en las medidas de higiene para mitigar la propagación de la infección, que puede transmitirse por el contacto directo con las secreciones oculares, por aerosoles (vía respiratoria) y las heces (vía fecal-oral).3,4
La conjuntivitis hemorrágica aguda causada por el viruas CVA24v se caracterizó por brotes grandes y explosivos desde su aparición en el año 1970. En la epidemiología del CVA24v se describen 4 etapas: la primera, la emergente, en la década de los años 70 y restringida al sudeste asiático y la India; la segunda, de expansión mundial en la década de los años 80, donde prácticamente comenzó el comportamiento pandémico; la tercera etapa, en la década de los años 90, con pocos reportes pero, evidente el comportamiento epidémico al menos en la región del Caribe y en países asiáticos; y la cuarta etapa, que comenzó a inicios del año 2000 y que se extiende hasta la actualidad con al menos 3 picos pandémicos (2000-2004, 2008-2010 y 2014-2017).5,7
Dichas etapas se hacen corresponder con los genotipos descritos en la epidemiología molecular de este agente, donde la región del genoma que codifica para la proteasa principal del virus (3C) es una de las regiones más estudiada por su conservación estructural y distintiva de las variantes circulantes. Sin embargo, en las últimas décadas, el análisis de la región (completa o parcial) de la proteína estructural VP1 ha tomado auge por su correspondencia taxonómica con los serotipos y genotipos (intra e inter serotipos) y contiene los sitios antigénicos principales de la cápside. Mediante estos estudios se describen 4 genotipos GI-GIV en base a la región del genoma 3C que se hacen corresponder con el análisis de la región estructural VP1, excepto el GII (posiblemente por la carencia de las secuencias de la región VP1 de este periodo).8-10
En la región de las Américas hasta el momento, solo investigadores brasileños han estudiado filogenéticamente las epidemias de conjuntivitis hemorrágica aguda, de forma similar que otros autores estudiaron las epidemias de los años 2003 y 2017 en Guyana Francesa y a nivel mundial solo se encuentra un estudio filodinámico desarrollado por investigadores de Taiwán en el 2016.7,10,11
En Cuba, la circulación del CVA24v coincide con sus etapas pandémicas. La primera epidemia causada por este agente ocurrió en el año 1986 y posteriormente ocurrieron otras epidemias significativas en los años 1987, 1992-1993, 1997, 2003, 2008-2009, y la última
Teniendo en cuenta este escenario y la disponibilidad de un número considerable de secuencias nucleotídicas del CVA24v a partir de los aislamientos virales obtenidos durante los primeros 5 periodos epidémicos cubanos (1986-2003), nos propusimos realizar una caracterización molecular basada en los análisis filogenéticos, filodinámicos, cambios aminoacídicos y análisis de secuencias obtenidas de cepas de muestras de heces, de las 2 regiones del genoma viral del CVA24v: la proteasa 3C y la proteína estructural VP1.
MÉTODOS
En esta investigación se realizaron 3 estudios descriptivos para identificar las variantes genéticas del CVA24v que circularon en Cuba durante los 5 periodos epidémicos de conjuntivitis hemorrágica aguda (1986-2009); inferir la filodinámica del CVA24v haciendo énfasis en las variantes que circularon en Cuba durante los 5 periodos epidémicos de conjuntivitis hemorrágica aguda (1986-2009) y analizar a nivel molecular las cepas cubanas del CVA24v obtenidas a partir de muestras clínicas de heces.
Muestras: El universo de estudio consistió de 172 secuencias obtenidas para la región genómica de la proteasa 3C y 149 secuencias obtenidas para la región genómica VP1 de cepas del CVA24v, aisladas durante los 5 periodos epidémicos de conjuntivitis hemorrágica aguda que ocurrieron en Cuba entre los años 1986 y el año 2009.
Análisis filogenético
Para los análisis filogenéticos y filodinámicos se seleccionó primeramente el modelo de sustitución nucleotídica a través del programa JModeltest v2.1.4.14 Posteriormente, las inferencias filogenéticas, así como los análisis filodinámicos se realizaron con los métodos bayesianos mediante el programa BEAST v1.8.4.15 En una primera etapa se generó el fichero de entrada al programa BEAST v1.8.4 a través del programa BEAUti incluido en el paquete de dicho programa.15
La selección de la combinación de los modelos de reloj molecular y los modelos epidemiológicos de los árboles a priori se realizó estimando la (log) verosimilitud marginal a través del método de path sampling (PS) descrito por Baele y colaboradores. El cálculo de la verosimilitud marginal (log likelihood) por PS se llevó a cabo en una primera etapa para modelos con reloj molecular estricto o relajado, empleando una distribución exponencial no correlacionada (UCED) o una distribución logaritmica normal no correlacionada (UCLD) combinados con el modelo de coalescencia no paramétrico Bayesian Skyride Plot (BSP).16
En todos los casos se llevaron a cabo 2 corridas de las cadenas de Markov y Monte Carlo (MCMC) del enfoque bayesiano, con 70 millones de generaciones para la región 3C y 100 millones para la región VP1, para lograr convergencia en los resultados y obtener un tamaño efectivo de la muestra superior a 200 en los parámetros de interés.17 Cada corrida se analizó individualmente y se combinó empleando como “burn-in” el 10 %, mediante el programa Tracer v1.7.1.18
Para el análisis de la distribución posterior de los árboles y la selección del árbol de mayor credibilidad, del inglés Maximum Clade Credidibility (MCC), se empleó el programa TreeAnnotator v1.8.4.19 Todos los árboles obtenidos se visualizaron a través del programa Figtree v1.4 disponible en el sitio.20
Las tasas de sustituciones por sitio por año y el tiempo del ancestro común más reciente del inglés most recent common ancestor (TMRCA) se estimaron empleando un enfoque bayesiano de cadenas de Markov y Monte Carlo (MCMC), así como la reconstrucción de la población viral y de la historia dinámica espacio-temporal sin grupo externo, con el BSP disponible en el BEAST.15
Para la filogeografía se utilizó como modelo de sustitución un modelo de difusión discreta donde los estados fueron los países usando una cadena de Markov continua tipo estándar. Para inferir la red social que indicó las rutas de propagación del virus se usó el método de selección de variables por búsqueda estocástica bayesiana, del inglés, Bayesian Stochastic Search Variable Selection, (BSSVS). Los resultados del análisis filogeográfico se visualizaron con el programa bayesiano SpreadD3, del inglés Spatial Phylogenetic Reconstruction of EvolutionAry Dynamics.21 Se realizó la prueba del Factor de Bayes (FB) para obtener los datos estadísticos que explicaran adecuadamente los procesos filogeográficos y se tomaron como significativos aquellos valores mayores que 3 (BF > 3).
A partir de las rutas de transmisión con BF > 3 se realizó además un estudio de redes de transmisión mediante el algoritmo Walktrap descrito por Pons y Lapaty.22) Determinación de los cambios aminoacídicos de las secuencias cubanas respecto a la cepa prototipo EH24_70_Singapur 1970 en las regiones genómicas parciales 3C y VP1. Los cambios aminoacídicos de las secuencias cubanas respecto a la cepa prototipo EH24_70_Singapur 1970 en las 2 regiones del genoma estudiadas se determinaron mediante el uso del programa MEGA6.23
En una primera aproximación en la búsqueda de la relevancia estructural y funcional de las posiciones mutadas en la región genómica 3C, se realizó un análisis en el sitio de internet Prosite utilizando como base la estructura de la proteasa 3C del CVA16 disponible.24) Teniendo en cuenta la disponibilidad de la estructura cristalina del CVA24v determinada por Zocher y colaboradores, se visualizaron esquemáticamente los cambios aminoacídicos encontrados en los 234 pb en la proteína VP1 mediante el programa DeepView 4.10.25
Análisis molecular de las cepas del CV-A24v cubanas obtenidas a partir de muestras clínicas de heces
El objetivo fundamental de esta tarea fue analizar a nivel molecular las 34 secuencias del CVA24v cubanas que se obtuvieron a partir de aislamientos de heces de pacientes con sospecha clínica de conjuntivitis hemorrágica aguda de los periodos epidémicos 1997, 2003, 2008-2009.
Las mismas se distribuyeron en los periodos epidémicos de la siguiente manera: 16 aislamientos del año 1997 obtenidos de 7 pacientes; 16 del año 2003 logrados de 16 pacientes y 2 del periodo 2008-2009, cada uno correspondiente a un paciente diferente en cada año. Los aislamientos a partir de las muestras clínicas de heces formaron parte de la muestra objeto de estudio según lo descrito en el acápite análisis filogenético. Es decir, que no hubo distinción en cuanto al tipo de muestra de la cual procedió la cepa viral para la realización de los análisis.
Mediante el programa Bioedit v7.0.5.326 se determinaron los porcentajes de identidad nucleotídica entre las secuencias obtenidas a partir de heces y las obtenidas a partir de los exudados, de diferentes pacientes y de un mismo paciente, en cada uno de los 3 periodos epidémicos analizados.
Consideraciones éticas de la investigación
La presente investigación se desarrolló de acuerdo a las normas establecidas en la Declaración de Helsinki (Asociación Médica Mundial, 2013).27 El protocolo de investigación se analizó por la Comisión Científica Especializada de Microbiología y por el Comité de Ética Institucional del IPK (CEI-IPK 06-17).
La presente investigación utilizó las secuencias obtenidas de los aislamientos virales conservados en congelación y obtenidos de muestras clínicas para diagnóstico tomadas de pacientes (niños y adultos) cubanos con cuadros clínicos de conjuntivitis hemorrágica aguda que se enviaron al laboratorio de Referencia de Enterovirus del IPK. Por lo tanto, el estudio no ofreció ningún riesgo ni daño ético para dichos individuos. En ningún momento se develó ningún dato identitario de los pacientes de los que procedían las muestras.
RESULTADOS
Conjuntos de alineamientos
En el presente trabajo los conjuntos de alineamientos sin secuencias dúplex, incluyeron 54 secuencias cubanas y 83 secuencias de 17 países para la región genómica 3C y 35 secuencias cubanas con 95 secuencias de 18 países para la región VP1. Las secuencias de los otros países se escogieron, de forma tal, que representaran tanto en tiempo como en regiones geográficas los periodos epidémicos en estudio.
En los conjuntos no se identificaron eventos de recombinación entre las secuencias seleccionadas, ni con los diversos serotipos de la especie C de los enterovirus en dichas regiones genómicas, a la que pertenece esta variante antigénica causante de la conjuntivitis hemorrágica aguda.
Caracterización filogenética
En el periodo epidémico de los años 1986-1987 las secuencias cubanas se agruparon con secuencias de Jamaica (1987), Brasil (1987) y África (1987) (figuras 1y2). En el periodo epidémico de los años 1992 y 1993 en la región genómica 3C, las secuencias cubanas mostraron homología con la única secuencia de la República Dominicana, en la región VP1 formaron una rama independiente. Ambos periodos epidémicos se incluyeron dentro del genotipo III (figuras 1y2). Para el periodo epidémico del año 1997 las secuencias cubanas mostraron relaciones de ancestralidad con una secuencia de los Estados Unidos en ambas filogenias. Sin embargo, en la región genómica VP1, se unió, además, a esta rama la secuencia dominicana del año 1993. Estas secuencias mostraron relaciones ancestrales más cercanas con las secuencias clasificadas dentro del genotipo GIV respecto al genotipo III (figuras 1y2). En el periodo epidémico del año 2003, las secuencias cubanas se dividieron en 2 grupos, uno mostró homología con las secuencias de Guadalupe en ambas inferencias, y el otro grupo con las secuencias de la India (2003) y Brasil (2003-2005) en la región del genoma 3C y con una secuencia de Guyana Francesa en la región genómica VP1 (figuras 1y2). El último periodo epidémico cubano (años 2008-2009) aparece solamente en el análisis de la región 3C y las secuencias cubanas mostraron relaciones de ancestralidad con secuencias de la India del año 2007 y un grupo de secuencias de Brasil del año 2009 (figuras 1y2).
 Fuente: Sci Rep 2020, 10, 13761.28
Fuente: Sci Rep 2020, 10, 13761.28
Fig. 1 Análisis filogenético de la región 3C (507 pb) de 54 secuencias de las epidemias cubanas de conjuntivitis hemorrágica aguda (1986-2009) y 82 secuencias de 17 países. Las barras azules en los nodos indican el 95 % HPD de los TMRCA. Los valores numéricos en los nodos indican la probabilidad posterior de la inferencia Bayesiana mayores de 0,700 (70,0 %). La longitud de las ramas indica el tiempo de evolución y la barra de la escala indica el tiempo calendario. Sobre las ramas principales se indican los genotipos para la región 3C (GI-GIV) según este estudio. Las secuencias se identificaron por su número de acceso al banco de genes GenBank, nombre, país y año de la cepa correspondiente. Resaltados en rojo los periodos epidémicos cubanos estudiados.
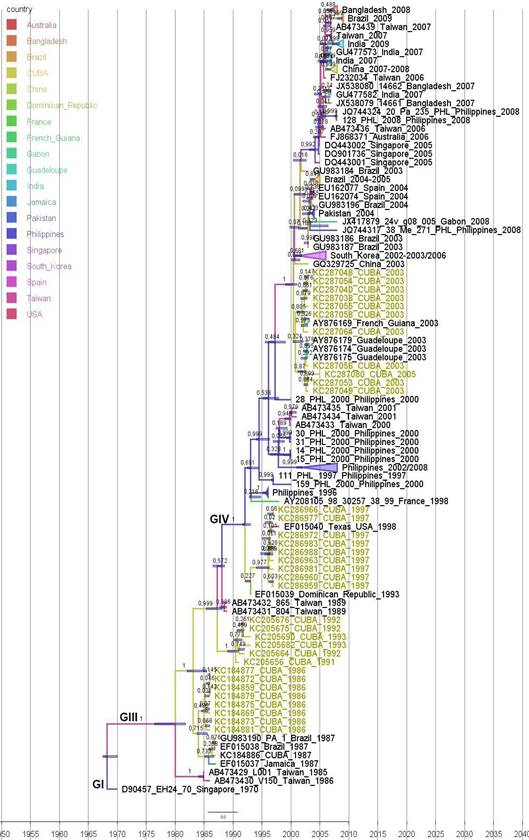 Fuente: Sci Rep 2020, 10, 13761.28
Fuente: Sci Rep 2020, 10, 13761.28
Fig. 2 Análisis filogenético de la región VP1 (234 pb) de 35 secuencias de las epidemias cubanas de conjuntivitis hemorrágica aguda (1986-2009) y 96 secuencias de 19 países. Las barras azules en los nodos indican el 95 % HPD de los TMRCA. Los valores numéricos en los nodos indican la probabilidad posterior de la inferencia Bayesiana mayores de 0,700. La longitud de las ramas indica el tiempo de evolución y la barra de la escala indica el tiempo calendario. Sobre las ramas principales se indican los genotipos para la región VP1 (GI, GIII, GIV) según este estudio. Las secuencias se identificaron por su número de acceso al banco de genes GenBank, nombre, país y año de la cepa correspondiente. Resaltados en colores los periodos epidémicos cubanos estudiados.
Análisis filodinámico
El análisis filodinámico de las variantes del CVA24v corroboró el origen monofilético de esta variante a partir de las que circularon en la primera epidemia en 1970. Se observó para ambas regiones genómicas, una topología poco equilibrada o no balanceada (similar a una escalera) lo que implica que ocurren cuellos de botella en la transmisión viral bajo la continua selección, dirigida por la inmunidad de rebaño, de forma que los linajes virales prevalentes existentes se reemplazan secuencialmente por nuevos grupos emergentes.
Este hecho se correspondió con los resultados del análisis mediante Skyride Bayesiano donde el virus mostró una población efectiva estable en el tiempo, solo con fluctuaciones (aumentos) correspondientes a los diferentes periodos pandémicos, con una tasa de sustitución nucleotídica (sustituciones/sitio/año) para la región 3C de 4,39 × 10-3 y para la región estructural VP1 de 5,80 × 10-3.
Las estimaciones del TMRCA mostraron que la evolución de nuevos genotipos ocurrió entre 4 a 5 años antes de su expansión global, y específicamente en Cuba las variantes pudieron estar circulando previamente entre 1 y hasta 2 años antes del desarrollo de los picos epidémicos (tabla 1).
Tabla 1 Rutas de conexiones epidemiológicas del CV-A24v durante el periodo 1986-2009 con BF > 3,0 en base a las regiones del genoma viral 3C y VP1
|
Región 3C Rutas de transmisión Desde-Hasta |
BF | Desde |
Región VP1 Rutas de transmisión Hasta |
BF | |
|---|---|---|---|---|---|
| Brasil | China | 11,1 | Australia | Singapur | 5,5 |
| Brasil | Jamaica | 11,4 | Australia | Taiwán | 5,9 |
| Brasil | Japón | 8,7 | Bangladesh | India | 17,6 |
| China | Estados Unidos | 43,9 | Bangladesh | Taiwán | 9,8 |
| China | Guadalupe | 28,3 | Brasil | China | 23,0 |
| China | Guyana Francesa | 7,9 | Brasil | Jamaica | 4,8 |
| China | India | 32,9 | Brasil | Pakistán | 16,4 |
| China | Jamaica | 23,6 | Brasil | Corea del Sur | 3,8 |
| China | Marruecos | 4,9 | Brasil | España | 7,8 |
| China | Tailandia | 3,5 | Brasil | Taiwán | 11,9 |
| Corea del Sur | Estados Unidos | 3,4 | China | Guadalupe | 38,6 |
| Corea del Sur | Taiwán | 3,0 | China | Guyana Francesa | 49,2 |
| Cuba | Taiwán | 161,7 | China | Jamaica | 8,8 |
| Estados Unidos | República Popular del Congo | 3,6 | China | Corea del Sur | 3,0 |
| Jamaica | Estados Unidos | 6,6 | China | Taiwán | 14,5 |
| República Dominicana | Singapur | 29,4 | Corea del Sur | Taiwán | 3,2 |
| Tailandia | Estados Unidos | 5,62 | Cuba | Taiwán | 19,4 |
| Taiwán | Estados Unidos | 432 | Francia | Filipinas | 7,2 |
| Taiwán | República Popular del Congo | 23,7 | Filipinas | Taiwán | 39,7 |
| Gabón | Pakistán | 3,3 | |||
| India | Taiwán | 13,7 | |||
| Pakistán | España | 9,7 | |||
| República Dominicana | Filipinas | 4,7 | |||
| República Dominicana | Taiwán | 3,1 | |||
| Singapur | Taiwán | 44,1 | |||
Fuente: Sci Rep 2020, 10, 13761.28
Predominaron las rutas de transmisión con origen en el continente asiático. China se destacó como una importante fuente de propagación de las variantes genéticas en Asia y Brasil en el continente americano.
Cuba no se identificó como el destino final de ninguna ruta de transmisión viral positiva (BF > 3) directa, pero basados en las relaciones filogenéticas de las secuencias cubanas con países destinos en algunas rutas obtenidas y el estudio mediante el programa Waltrap, se propusieron posibles rutas de introducción del virus al país (figura 3). Se identificó la ruta China-Jamaica para el periodo epidémico 1986-1987 obtenida en ambas regiones genómicas (3C y VP1). Para el periodo epidémico del año 1997 se identificaron varios países de origen (basados en la región 3C): China, Corea del Sur, Tailandia y Taiwán, todas con destino en los Estados Unidos. Para el periodo epidémico del año 2003 se identificaron dos rutas de transmisión viral en ambas regiones genómicas: desde China hasta Guadalupe y Guyana Francesa. En el periodo 2008-2009 se obtuvo una ruta desde China a la India en la región 3C.
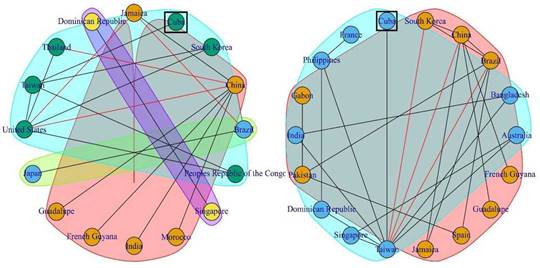 Fuente: Sci Rep 2020, 10, 13761.28
Fuente: Sci Rep 2020, 10, 13761.28
Fig. 3 Redes de transmisión viral obtenidas mediante el análisis Walktrap a partir de los valores de BF > 3 para las regiones del genoma 3C (izquierda) y VP1 (derecha). Enmarcada en cuadrado verde la posición de Cuba.
Cambios amonoacídicos
La representación estructural de los cambios aminoacídicos en la proteasa 3C de las secuencias cubanas los situó mayoritariamente, dentro o cercanos, al sitio catalítico de la enzima o al sitio de unión al oriL (vital en el inicio de la replicación viral) (figura 4A y B).
Por otro lado, se observó la composición conservada de determinadas posiciones en la región 3C, claves en el funcionamiento de esta proteasa y que han sido objetivos para la búsqueda y el diseño de nuevos antivirales enterovirales.29,30
Los cambios aminoacídicos de las cepas cubanas en la proteína VP1 se localizaron su mayoría, en la región del cañón (figura 4C).

Fig. 4 A y B: Representación estructural del centro catalítico (A) y el sitio de unión al oriL (B) con los cambios aminoacídicos de las secuencias cubanas del CVA24v en la proteasa 3C, en la estructura de la proteasa 3C del CVA16 (entrada PDB 3SJI). Se resaltan en rojo el sitio catalítico, en amarillo el sitio de unión al oriL, en verde las posiciones mutantes, y en azul las posiciones potencialmente relevantes desde el punto de vista funcional. C: Cambios aminoacídicos en la región genómica VP1 de las secuencias cubanas (círculos azules aqua) representados en la vista lateral de un protómero, VP1 (amarillo), VP2 (azul), VP3 (verde), VP4 (rojo) proteínas estructurales de la cápside.
Análisis de las secuencias obtenidas de aislamientos de heces
Las secuencias obtenidas de los aislamientos de las heces mostraron porcentajes de identidad nucleotídica de 97,7 %-100 % y 97,3 %-100 %, respecto a las secuencias de los aislamientos obtenidos de los exudados en las regiones genómicas 3C y VP1 respectivamente.
Se observaron, además 3 cambios aminoacídicos en la región 3C y 1 en la región VP1 en las secuencias de 7 cepas seriadas de un paciente aisladas en 1 periodo de 20 días.
Por otra parte, en los diferentes análisis tanto filogenéticos como filodinámicos, las secuencias nucleotídicas de los virus obtenidos a partir de las heces se comportaron tal cual las de los exudados conjuntivales en el grupo de secuencias de un mismo periodo epidémico.
DISCUSIÓN
Cuba ha sido un área activa de circulación del CVA24v en las Américas desde mediados de la década de 1980.12,13 La caracterización filogenética de ambas regiones del genoma viral mediante el programa BEAST, determinó que cada periodo epidémico cubano ocurrió cíclicamente entre 5 a 6 años resultado de una nueva introducción del virus al país y no de la circulación o emergencia endémica de una variante genética y en los años epidémicos continuos (generalmente 2 años) circuló la misma variante genética. Los brotes cubanos de conjuntivitis hemorrágica aguda en los periodos estudiados, fueron causados por variantes genéticas del CV-A24v homólogas a las que circularon en la región de las Américas en cada uno de ellos. En algunos periodos epidémicos (1986-1987, 2003 y 2008-2009), se pudo evidenciar las relaciones de ancestralidad con secuencias de otros continentes (África y Asia), lo que avala también el alto potencial epidémico de esta variante antigénica.
Los resultados indicaron, además, que para cada periodo epidémico el CVA24v pudo estar circulando de 1 año a 2 años previos, a que ocurrieran los picos epidémicos. Estos resultados apoyan las observaciones epidemiológicas y del laboratorio donde en algunos de estos periodos epidémicos cubanos previos a los años epidémicos se reportaron y diagnosticaron casos con conjuntivitis hemorrágica aguda. Este hallazgo pudiera tenerse en cuenta para futuros eventos epidémicos de esta variante antigénica, lo cual tributaría en una alerta oportuna al sistema nacional de salud.
En los últimos años ha existido mucha controversia sobre qué región del genoma es más informativa desde el punto de vista filogenético.6,31,32 Los primeros estudios filogenéticos del CVA24v se basaron en la región 3C no estructural, sin embargo, el análisis reciente de esta región genómica ha sido reemplazado por el análisis de la región estructural VP1.11,8,31 En este trabajo, ambas regiones se mostraron igualmente informativas para los análisis filogenéticos y filodinámicos, de modo que es necesario llegar a un consenso respecto a qué región genómica analizar y qué secuencias de referencia se pueden elegir por períodos y origen geográfico para obtener una mejor clasificación y análisis de la epidemiología molecular del CVA24v.
El análisis filogenético reveló un desarrollo secuencial de los genotipos, con cepas pertenecientes al genotipo IV aisladas durante el brote de AHC en Cuba en 1997. Comportamiento semejante lo mostraron las cepas aisladas en 1996 en Filipinas y en Francia y los Estados Unidos en 1998, lo que indica una ocurrencia anterior a mediados de la década de 1990 y no a principios del 2000 del genotipo IV. Otros estudios mostraron una topología similar a los árboles filogenéticos obtenidos, incluso con modelos menos robustos.5,10,25,33-38 La definición de genotipo o subgenotipo del CVA24v no se ha establecido claramente. Varios estudios han demostrado diferencias espacio-temporal entre las cepas de diferentes países mediante el uso de las regiones genómicas 3C y VP1 o del genoma completo.11,33,34-36
Los estudios filogenéticos mostraron un reemplazo cronológico alternativo de secuencias latinoamericanas y europeas, con secuencias asiáticas al igual que las encontradas por otros autores.6,11,22,37 Además, en los estudios filogeográficos mostraron a Asia como el principal generador de rutas de transmisión así la existencia de numerosas rutas hacia ese continente. Cabría preguntarse la posibilidad de que, a partir de la primera oleada pandémica, las variantes genéticas en su recorrido por el mundo evolucionan de forma tal que cuando retornan al sudeste asiático, constituyen nuevas variantes que, en medio de poblaciones susceptibles, encuentran todas las condiciones para originar otra oleada pandémica.
A partir del estudio de los cambios aminoacídicos de las cepas cubanas se encontró que la mayoría de los mismos en la proteína VP1, se localizaron en la región del cañón. En esta región se encuentran los sitios antigénicos y, por ende, dichos cambios pudieran permitir el escape a la acción neutralizante de los anticuerpos generados por variantes virales que circularon en las epidemias previas. En la región 3C, los cambios aminoacídicos se localizaron cerca del sitio catalítico de la enzima o de unión al oriL de forma que pudiera estar involucrados en la evolución de las variantes virales cubanas. Por otro lado, se observó la composición conservada de determinadas posiciones, claves en el funcionamiento de esta proteasa, lo que contribuye a la inclusión de esta variante viral en el diseño y búsqueda de nuevos antivirales dirigidos contra esta proteína viral en los enterovirus.29,30
También se demostró a nivel molecular, que, en efecto, la variante genética del CVA24v circulante causante de la conjuntivitis hemorrágica aguda es capaz de replicarse en la conjuntiva y el intestino de los pacientes y excretarse en sus heces. En los diferentes análisis tanto filogenéticos como filodinámicos, las secuencias nucleotídicas de los virus obtenidos a partir de las heces se comportaron tal cual las de los exudados conjuntivales en el grupo de secuencias de un mismo periodo epidémico. Precisamente, este hecho nos permitió argumentar algunos aspectos epidemiológicos y de la patogenia de esta variante viral que han sido poco difundidos y estudiados en la comunidad científica y que parten del hecho de analizar secuencias del CVA24v obtenidas de aislamientos de heces. En consecuencia, se incluyeron secuencias de CVA24v obtenidas de heces de casos de parálisis fláccida aguda (PFA) en Filipinas (n = 13), India (n = 4), Bangladesh (n = 9) y Gabón (n = 1), de primates no humanos (n = 3, Bangladesh) y de muestras ambientales (n = 5, Filipinas).37-42 Todas las secuencias de CVA24v se agruparon en los 3 genotipos principales dentro de la región VP1, independientemente del tipo de muestra de la que se obtuvieron los aislamientos.
De modo que estos resultados argumentan: a) el tropismo neurológico del CVA24v, a tener en cuenta por su alto potencial epidémico y pandémico; b) el rango de hospederos de esta variante antigénica, desde los individuos asintomáticos, con PFA y primates no humanos que pueden servir de reservorios temporales; y c) la permanencia en el medio ambiente (aguas albañales, ríos y afluentes) que sirven de vehículos para la diseminación viral, ya sea, entre, durante y años posteriores al desarrollo de las epidemias.
Conclusiones
Este estudio es uno de los estudios más completos realizados en las Américas sobre el origen, evolución y rutas de transmisión de CVA24v en la región. Los hallazgos resuelven una pregunta de larga data sobre cuándo y dónde podrían originarse las epidemias CVA24v en Cuba. La detección inesperada de cepas de CVA24v pertenecientes al genotipo IV a mediados de la década de 1990 destaca la necesidad de revisar el origen del genotipo IV. Además, subrayan la importancia de comprender la evolución global de CVA24 y su amenaza pandémica. Por otro lado, el estudio enfatiza la importancia de las heces como una ruta adicional de transmisión del CVA24v y argumenta el espectro diverso de síndromes clínicos causados por la infección por CVA24v y de otros reservorios no humanos.
Los resultados del estudio no solo arrojan luz sobre la diversidad genética, la evolución y la transmisión global de CVA24v, sino que también pueden ayudar a identificar nuevas estrategias de control para futuras epidemias a nivel nacional, regional y mundial.

