










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Estomatología
versión On-line ISSN 1561-297X
Rev Cubana Estomatol vol.52 no.2 Ciudad de La Habana abr.-jun. 2015
PRESENTACIÓN DE CASO
Síndrome Gorlin-Goltz asociado a fisura labiopalatina bilateral
Gorlin-Goltz syndrome associated with bilateral cleft lip and palate
Noemí Lorena Leiva Villagra, Sebastián Alejandro Véliz Méndez, Leonardo Esteban González Escobar, Carolina Andrea Salazar Ponce
Facultad de Odontología. Universidad de Chile. Santiago de Chile.
RESUMEN
El síndrome de Gorlin-Goltz corresponde a un trastorno de herencia autosómica dominante. Uno de los criterios menores de este síndrome es la fisura labiopalatina. Si bien esta corresponde a la anomalía congénita maxilofacial más prevalente, un porcentaje variable está asociado a síndromes. Presentar un caso de un paciente con síndrome de Gorlin-Goltz y fisura labiopalatina bilateral asociada constituye el propósito de esta presentación. Se trata de un paciente de 12 años de edad, con diagnóstico de síndrome de Gorlin-Goltz remitido por genetista. Clínicamente presenta anomalías cutáneas, óseas, dentarias, neurológicas, tumores, hoyuelos palmoplantares, prognatismo mandibular y fisura labiopalatina bilateral operada. Es importante reconocer las características asociadas no solo al área craneofacial, sino también a otras partes del cuerpo. Se requiere de la atención de un equipo multidisciplinario en el que el odontólogo también debe participar. La mayoría de las publicaciones se enfocan solo en el manejo quirúrgico de los quistes y no en el de otras secuelas asociadas, como lo es la fisura labiopalatina.
Palabras clave: síndrome Gorlin-Goltz, fisura labiopalatina, fisura labiopalatina sindrómica.
ABSTRACT
Gorlin-Goltz syndrome is an autosomal dominant inheritance disorder. Cleft lip and palate is one of the minor criteria for this syndrome. Cleft lip and palate is the most prevalent congenital maxillofacial anomaly, and a varying percentage is associated with syndromes. Present the case of a patient with Gorlin-Goltz syndrome and associated bilateral cleft lip and palate. A 12-year-old male patient was referred by the geneticist with a diagnosis of Gorlin-Goltz syndrome. Clinical examination revealed skin, bone, dental and neurological anomalies, as well as tumors, palmoplantar pits, mandibular prognathism and operated bilateral cleft lip and palate. It is important to examine not only features associated with the craniofacial region, but also with other parts of the body. An interdisciplinary team is required of which the odontologist should be a member. Most publications only refer to the surgical management of cysts and not to associated sequels, such as cleft lip and palate.
Key words: Gorlin-Goltz syndrome, cleft lip and palate, syndromic cleft lip and palate.
INTRODUCCIÓN
El síndrome de Gorlin-Goltz (SGG), también conocido como síndrome nevus de células basales (SNCB) es un trastorno de herencia autosómica dominante en 60 % y 80 % de los casos,1-3 los que presentan penetrancia elevada y expresión fenotípica variable. Se relaciona con una alteración a nivel del cromosoma 9 (9q22.3-q31). La mutación principal ocurre en el gen PATCHED (PTCH)4 del cromosoma 9q, el cual, entre otras funciones actúa como un gen supresor de tumores.1,5,6 Entre el 35 % y 50 % de los casos la mutación no se hereda, y aparece como una mutación espontánea.
Desde el punto de vista clínico este síndrome es muy complejo e incluye una gran variedad de anomalías cutáneas, dentales, óseas, oftálmicas, neurológicas y sexuales.7 Los carcinomas nevus basocelulares (CNVB) son la principal lesión cutánea, la cual se manifiesta entre la pubertad y los 35 años de edad.8 Se ubica en las zonas torácicas y cervicofacial. Otras alteraciones cutáneas encontradas en estos pacientes son los quistes sebáceos9 y tumores cutáneos plantares o palmares.8 A nivel torácico la alteración más común es la presencia de costillas bífidas o fusionadas (40 % de los pacientes).10 A nivel vertebral pueden presentar espina bífida oculta, cifoescoliosis y hemivertebras. Las anomalías neurológicas son retardo mental, calcificación dural,11 génesis del cuerpo calloso, hidrocefalia, meduloblastomas. A nivel sexual, la anomalía más prevalente en el sexo masculino es el hipogonadismo, mientras que en el sexo femenino son los tumores de ovario.
Los tumores odontogénicos queratoquísticos (TOQ), ocurren en casi 75 % de los pacientes que presentan el síndrome y pueden ser el primer signo de la enfermedad.12 Suele aparecer durante la primera década de vida,1,8 generalmente como un hallazgo radiográfico. Casi 70 % de los pacientes con el SCNBC presentan algún grado de alteraciones cráneo-faciales.8,13,14 Estas alteraciones comprenden frente amplia y alta, hueso frontal y parietales abombados, arcos supraciliares altos, puente nasal ensanchado asociado o no a hipertelorismo.5,15,16 El maxilar puede ser hipoplásico y la mandíbula hiperplásica con un prognatismo variable. Aunque menos comunes, estos pacientes pueden presentar paladar profundo, labio y paladar hendidos, agenesias dentarias. La fisura labio palatina (FLP) es relativamente poco frecuente en el SGG. Kimonis y otros,15 analizaron a 105 pacientes con SGG; solo en 3 de ellos hallaron fisura de labio o paladar. Un porcentaje variable del total de fisuras labiopalatinas está asociada a síndromes. Estudios recientes han mostrado prevalencia entre 6 % y 14 % de FLP sindrómica.17,18
Debido a lo poca frecuencia y escasa publicación de casos como este, el objetivo de este artículo es reportar un caso de síndrome de Gorlin-Goltz con fisura labiopalatina bilateral.
PRESENTACIÓN DEL CASO
Paciente de 12 años de edad, sexo masculino, derivado a la Unidad de Malformación Craneofacial con diagnóstico de síndrome de Gorlin Goltz.
Anamnesis: Paciente hijo único de padres no consanguíneos. Embarazo normal sin contacto con teratógenos, ecografías prenatales mostraron macrocefalia. Parto de término por cesárea a las 39 semanas de gestación, con un peso de 3,920 g, 51,5 cm de longitud, y perímetro craneal al nacer de 40 cm (> P98). Al nacer su madre tenía 30 años de edad y su padre 35 años, no hay antecedentes de casos similares en su familia. Tuvo una alimentación a través de mamadera, sin succión de pecho materno a pesar de que la madre lo intentó.
Diagnósticos de recién nacido: Recién nacido a término adecuado para edad gestacional, síndrome malformativo, macrocefalia, fisura labiopalatina bilateral.
El paciente ha sido estudiado en la Unidad de Genética con un diagnóstico de síndrome de Gorlin-Goltz. Presentaba macrocefalia con dilatación del espacio subaracnoideo, hidrocefalia tratada a los 5 años mediante descompresión craneana y válvula ventrículo peritoneal, sin signos clínicos de mal funcionamiento. Malformación de Chiari tipo I, ventriculomegalia supratentorial, múltiples lesiones quísticas en huesos faciales, operados en dos oportunidades, fisura labiopalatina bilateral operada, otitis crónica tratada con miringotomía (instalación de colleras). Sostén cefálico a los 4 meses, marcha a los 2 años y 3 meses, lenguaje antes del año con rinolalia. Estudia en colegio normal con buen rendimiento escolar, talla normal alta, peso adecuado.
Examen físico: Presenta tórax asimétrico, abdomen blando, sin visceromegalia, escoliosis y dorso curvo, caderas de altura asimétrica, con un trastorno secundario de la marcha, extremidades finas y asimétricas, dedos cortos aguzados, pierna izquierda más corta, operado de ambas piernas a nivel de los tendones de Aquiles en septiembre de 2008 con franca mejoría en la movilidad y disminución de las contracturas, pie equino bilateral, lesión nodular en piel de palma izquierda, planta izquierda y tobillo derecho.
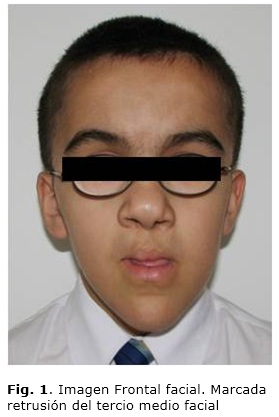
A nivel facial el paciente presenta facies alargada, frente amplia, tercio medio disminuido, nariz de base ancha y bulbosa, columela corta, labio leporino bilateral superior operado con cicatriz retráctil, dificultad en el cierre bucal, asimetría facial, desviación mandibular a la izquierda. En forma lateral presenta un perfil posterior anteinclinado, labio superior plano, corto e inferior evertido, surco mentolabial disminuido, falta de desarrollo del maxilar superior severa, asociado a una clase III esqueletal. A nivel ocular presenta miopía, estrabismo e hipermetropía severa con uso de lentes óptico (Fig. 1).
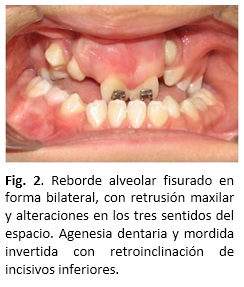
Examen clínico intraoral: Presenta vestíbulo superior y paladar duro operado con fisura residual bilateral, lengua de tamaño normal, pero no funcional, descendida en reposo, interpuesta en deglución y en fonación. La oclusión presenta en sentido sagital: mesioclusión molar bilateral franca, mordida invertida anterior severa, el resalte negativo severo, en sentido transversal: mordida cruzada bilateral franca, línea media superior desviada a la izquierda en 1,5 mm, y en sentido vertical: sobrepase (overbite) aumentado en 6 mm (Fig. 2).
Exámenes complementarios:
a) En la ortopantomografía se observó:
— Maxilar: Fisura palatina bilateral. No se observan gérmenes de dientes 1,8 y 2,8; dientes 1,3 y 2,2 ausentes; dientes 1,7, 1,5, 2,3, 2,4, 2,5, 2,7 en evolución intraósea; dientes 1,7 y 1,4 desplazadas, proyectados en seno maxilar derecho; diente 1,7 en posición levemente superior y mesial y diente 1,5 en posición distal con mesioversión franca. Se observa germen dentario supernumerario, en etapa de formación coronaria, proyectado a nivel de diente 1,1, la cual presenta giroversión.
— Mandíbula: No se observan gérmenes de dientes 4,8 ni 3,8; dientes 4,7, 4,3 y 3,7 ausentes. Diente 3,3 espacio pericoronario ensanchado; diente 3,5 en posición baja y levemente mesial; diente 4,2 leve giroversión.
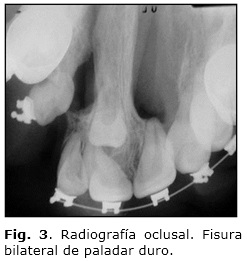
b) Radiografía oclusal: Se observa la continuidad de la fisura bilateral en el paladar, así como la presencia de dientes incluidos en este (Fig. 3).
El paciente fue diagnosticado con una lesión quística en el maxilar superior derecho en marzo del 2008 (quiste dentígero). Mediante la tomografía computada se observó que medía 46 × 48 × 57 mm en sus diámetros céfalo-caudal, transversal y anteroposterior, respectivamente. Esta lesión era homogénea, con contenido de densidad de partes blandas, que produjo importante remodelación de todas las paredes del seno maxilar derecho, en el cual se observaba colapsado hacia posterior. Había elevación del piso de la órbita derecha y desplazamiento medial de la pared lateral de la fosa nasal derecha, la cual se observaba ocluída en su porción anterior. Presentaba dos dientes incluidos, con reabsorción de las raíces dentarias de premolares y molares adyacentes al quiste. Fue operado días después en marzo de 2008.
Actualmente el paciente presenta senos frontales, maxilar izquierdo, etmoidales y esfenoidal de adecuado desarrollo y neumatización, con unidades de drenaje permeables, desviación izquierda del tabique nasal, que contacta con el cornete medio y estrecha la fosa nasal izquierda. Muestra, deformidad y falta de fusión ósea del paladar duro, compatible con diagnóstico de referencia.
A nivel de su columna presenta escoliosis dorsolumbar dextroconvexa, de amplio radio de curvatura con leve rotación de varios cuerpos vertebrales, pero con altura conservada, y muro posterior bien alineado. Conserva la altura de los espacios intervertebrales dorsales y lumbares. La cifosis dorsal y lordosis lumbosacra no presentaban variación radiológica significativa. Presentó un catéter de descarga de líquido cefalorraquídeo. Su pelvis presenta un mínimo desnivel respecto a la columna.
DISCUSIÓN
El síndrome de Gorlin-Goltz o síndrome nevus de células basales es una enfermedad poco frecuente. Se estima que tiene una incidencia de 1 por cada 60 000 individuos, de los cuales entre 35 % y 50 % de los casos corresponden a mutaciones espontáneas19 como lo es el caso presentado. El diagnóstico del SNCB se basa en criterios clínicos mayores y menores. Como criterios mayores, nuestro paciente presenta quiste dentígero en maxilar superior, hoyuelo palmar. Además, encontramos los siguientes criterios menores: frente amplia y levemente abombada, puente nasal ensanchado. Otra característica era la fisura palatina bilateral, retenciones dentarias, giro versiones dentarias y cifoescoliosis.
Debido a la alta variabilidad de la presentación clínica del síndrome, el paciente debe recibir atención y tratamiento por diferentes especialidades, tanto para determinar el diagnóstico, pronosticar el patrón genético y tratar las diversas manifestaciones clínicas. El manejo de estos pacientes dependerá de la alteración que se deba tratar. El tratamiento de tumores odontogénicos queratoquísticos asociados al síndrome no difieren de los no sindrómicos.8 Son fundamentales el seguimiento y los controles radiográficos periódicos, pues se sabe que los TOQ en los pacientes con el SNCB tienen mayor recurrencia que los no sindrómicos.13,14
La FLP es relativamente poco frecuente en el SGG. La mayoría de las publicaciones de casos hacen referencia al tratamiento quirúrgico de los tumores. Son pocas las publicaciones que hacen referencia explícita a esta asociación. Kimonis y otros15 analizaron a 105 pacientes con SGG, y hallaron que solo 3 de ellos presentaban fisura de labio o paladar. En 1997, Lambrecht y Kreusch publicaron una revisión de esta asociación, que de 719 pacientes publicados con SGG hasta esa fecha, 8,5 % (61 casos) presentaban fisuras orofaciales, postulando que la incidencia de FLP en pacientes con el síndrome es mayor a la establecida por Gorlin.
Es fundamental el tratamiento interdisciplinario en estos pacientes, involucrando especialidades médicas y odontológicas, así como otras áreas de la salud; con el fin de rehabilitar en forma integral a estos pacientes y asesorar a los padres durante los diversos procesos que deberá vivir el paciente.20 Es por ello que se hace necesario que toda persona que trabaje con pacientes con SGG conozcan en qué momento los especialistas deben realizar las evaluaciones.
REFERENCIAS BIBLIOGRÁFICAS
1. Ortega García de Amezaga A, García Arregui O, Zepeda Nuno S, Acha Sagredo A, Aguirre Urizar JM. Gorlin-Goltz syndrome: clinicopathologic aspects. Med Oral Patol Oral Cir Bucal. 2008;13(6):E338-43.
2. Kolokythas A, Fernandes RP, Pazoki A, Ord RA. Odontogenic keratocyst: to decompress or not to decompress? A comparative study of decompression and enucleation versus resection/peripheral ostectomy. J Oral Maxillofac Surg. 2007;65(4):640-4.
3. Rosón-Gómez S, González-García R, Naval-Gías L, Sastre-Pérez J, Muñoz-Guerra MF, Díaz-González FJ. Síndrome de Gorlin-Goltz: Serie de 7 casos. Rev Esp Cir Oral Maxilofac. 2009;31:309-15.
4. Lam C, Ou JC, Billingsley EM. “PTCH”-ing It Together: A Basal Cell Nevus Syndrome Review. Dermatol Surg. 2013;39(11):1557-72.
5. Snoeckx A, Vanhoenacker FM, Verhaert K, Chappelle K, Parizel PM. Gorlin-Goltz syndrome in a child: case report and clinical review. Jbr Btr. 2008;91(6):235-9.
6. Roncales-Samanes P, Pena-Segura JL, Fernando-Martinez R, Fuertes-Rodrigo C, Garcia-Oguiza A, Lopez-Pison J. Gorlin syndrome in the paediatric age. Rev Neurol. 2014;58(7):303-7.
7. Bree AF, Shah MR. Consensus statement from the first international colloquium on basal cell nevus syndrome (BCNS). Am J Med Genet A. 2011;155A(9):2091-7.
8. Kalogeropoulou C, Zampakis P, Kazantzi S, Kraniotis P, Mastronikolis NS. Gorlin-Goltz syndrome: incidental finding on routine ct scan following car accident. Cases J. 2009;2(9087):1757-626.
9. Feito-Rodriguez M, Sendagorta-Cudos E, Moratinos-Martinez M, Gonzalez-Beato MJ, de Lucas-Laguna R, Pizarro A. Dermatoscopic characteristics of acrochordon-like basal cell carcinomas in Gorlin-Goltz syndrome. J Am Acad Dermatol. 2009;60(5):857-61.
10. Agrawal A, Murari A, Vutukuri S, Singh A. Gorlin-goltz syndrome: case report of a rare hereditary disorder. Case Rep Dent. 2012;475439(10):23.
11. Saulite I, Voykov B, Mehra T, Hoetzenecker W, Guenova E. Incidental finding of lamellar calcification of the falx cerebri leading to the diagnosis of gorlin-goltz syndrome. Case Rep Dermatol. 2013;5(3):301-3.
12. Antonoglou GN, Sandor GK, Koidou VP, Papageorgiou SN. Non-syndromic and syndromic keratocystic odontogenic tumors: Systematic review and meta-analysis of recurrences. J Craniomaxillofac Surg. 2014 Oct;42(7):e364-71.
13. Longobardi G, Diana G, Poddi V, Pagano I. Follicular cyst of the jaw developing into a keratocyst in a patient with unrecognized Gorlin-Goltz syndrome. J Craniofac Surg. 2010;21(3):833-6.
14. Wilson C, Murphy M. Conservative management of multiple keratocystic odontogenic tumours in a child with Gorlin-Goltz syndrome: a case report. Eur J Paediatr Dent. 2008;9(4):195-8.
15. Kimonis VE, Goldstein AM, Pastakia B, Yang ML, Kase R, DiGiovanna JJ, et al. Clinical manifestations in 105 persons with nevoid basal cell carcinoma syndrome. Am J Med Genet. 1997;69(3):299-308.
16. Dixit S, Acharya S, Dixit PB. Multiple odontogenic keratocysts associated with Gorlin-Goltz syndrome. Kathmandu Univ Med J. 2009;7(28):414-8.
17. Doray B, Badila-Timbolschi D, Schaefer E, Fattori D, Monga B, Dott B, et al. [Epidemiology of orofacial clefts (1995-2006) in France (Congenital Malformations of Alsace Registry)]. Arch Pediatr. 2012;19(10):1021-9.
18. McDonnell R, Owens M, Delany C, Earley M, McGillivary A, Orr DJ, et-al. Epidemiology of Orofacial Clefts in the East of Ireland in the 25-Year Period 1984-2008. Cleft Palate Craniofac J. 2014;51(4):e63-9.
19. Gorlin RJ. Nevoid basal cell carcinoma (Gorlin) syndrome: unanswered issues. J Lab Clin Med. 1999;134(6):551-2.
20. Kimonis VE, Singh KE, Zhong R, Pastakia B, Digiovanna JJ, Bale SJ. Clinical and radiological features in young individuals with nevoid basal cell carcinoma syndrome. Genet Med. 2013;15(1):79-83.
Recibido: 26 de abril de 2014.
Aprobado: 29 de enero de 2015.
Noemí Leiva Villagra. Facultad de Odontología. Universidad de Chile. Santiago de Chile.
Correo electrónico: leivanoemi@yahoo.com

