










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Archivo Médico de Camagüey
versión On-line ISSN 1025-0255
AMC v.14 n.1 Camagüey ene.-feb. 2010
CASOS CLÍNICOS
Síndrome 48, XXXY: presentación de un caso
48, XXXY syndrome: a case report
Dra. Carmen María Chiong QuesadaI; Lic. Nelson H. Martín CuestaII; Dr. Roberto Hernández HernándezIII; Dr. Martín Chiong QuesadaIV
I Especialista de I Grado en Genética Clínica. Profesor Instructor. Centro Provincial de Genética. Camagüey, Cuba.
II Licenciado en Ciencias Biológicas. Profesor Instructor.
III Especialista de I Grado en Medicina General Integral. Profesor Instructor. Universidad de Ciencias Médicas. Camagüey, Cuba.
IV Especialista de I Grado en Gastroenterología. Hospital Provincial Clínico Quirúrgico Docente “Amalia Simoni”. Camagüey, Cuba
RESUMEN
Se presentó un paciente blanco, masculino, de 21 años de edad con dismorfia facial, hipogenitalismo, anomalías esqueléticas y retraso mental severo. Para el diagnóstico de esta afección se aplicó el método clínico a través de la técnica comparativa o de patrón. Se realizó estudio cromosómico en sangre periférica, se emplearon técnicas convencionales (banda GTG) confirmándose el diagnóstico del Síndrome 48, XXXY. Resulta de gran importancia tener en cuenta las manifestaciones clínicas antes mencionadas para establecer el diagnóstico precoz de este síndrome, ofrecer asesoramiento genético oportuno a los padres, así como rehabilitar física, psíquica y socialmente a estos pacientes.
DeCs: asesoramiento genético, síndrome de Klinefelter/ diagnósticoABSTRACT
A 21 years old masculine white patient with facial dysmorphia, hypogenitalism, skeletal anomalies and severe mental delay was presented. For the diagnosis of this affection the clinical method through the comparative or pattern technique was applied. The chromosomal study was carried out in peripheral blood, conventional techniques were used being confirmed the diagnosis of the 48, XXXY Syndrome. It is important to take into account the clinical manifestations before mentioned in order to establish early diagnosis, rehabilitate these patients physically – psychically and socially and to offer a genetic counselling to the family.
DeCS: genetic counseling; Klinefelter Syndrome/ diagnosis
INTRODUCCIÓN
En 1942 Harris Klinefelter1 publicó un reporte de nueve hombres con pechos desarrollados, vello escaso en cara y cuerpo, testes pequeños e incapacidad para producir espermatozoides, conociéndose a finales de los años 50 que estos individuos presentaban un cromosoma sexual adicional (47, XXY). Posteriormente se describen otras variantes que incluyeron dos e incluso tres cromosomas supernumerarios.1
El síndrome 48, XXXY es una variante del Síndrome Klinefelter, en el que existen dos cromosomas sexuales extra.2 Este desorden genético poco frecuente (1/50 000) suele aparecer como un fenómeno aislado. Según Smith3 se caracteriza por hipogenitalismo, pronación limitada del codo, conteo bajo de crestas dermales en la punta de los dedos y retraso mental de grado variable, cuadro provocado por un fallo doble en la disyunción cromosómica normal que ocurre durante la gametogénesis parental.2,3
Resulta de gran interés presentar este caso a todos lo médicos con el propósito de dar a conocer las principales manifestaciones clínicas de esta anomalía, que interesa los cromosomas sexuales. Se deriva la importancia de realizar un diagnóstico precoz, haciendo posible la evaluación e intervención psicopedagógica de estos pacientes, administrar terapia hormonal sustitutiva y ofrecer asesoramiento genético a los padres, logrando así mejorar significativamente el pronóstico de sus discapacidades.
CASO CLÍNICO
Paciente de 21 años de edad, raza blanca, sexo masculino, tercer hijo de una pareja de 29 y 27 años, la madre y el padre respectivamente, sanos y sin antecedentes patológicos familiares de interés.
Antecedentes prenatales: E3, P3, A0. Anemia en el segundo trimestre e infecciones urinarias en el segundo y tercer trimestre. Bajo peso materno.
Este paciente nació a las 40 semanas de gestación, de un parto distócico, con un peso de 1900 g, talla: 46cm, apgar 6/7, llanto demorado, depresión moderada, requiriendo la administración de oxígeno. Durante sus primeros años de vida presentó escasa ganancia de peso, llegando a ser un niño malnutrido por defecto (baja talla y bajo peso para su edad), con retardo en la erupción dentaria, en el desarrollo psicomotor y en el lenguaje. Requirió varios ingresos por procesos respiratorios recurrentes, disfunción tiroidea, hematurias sin causa aparente y luxación espontánea del codo, llevando seguimiento por varias especialidades. No recibió tratamiento sustitutivo hormonal requerido por el hipogonadismo hipergonadotrópico que presenta.
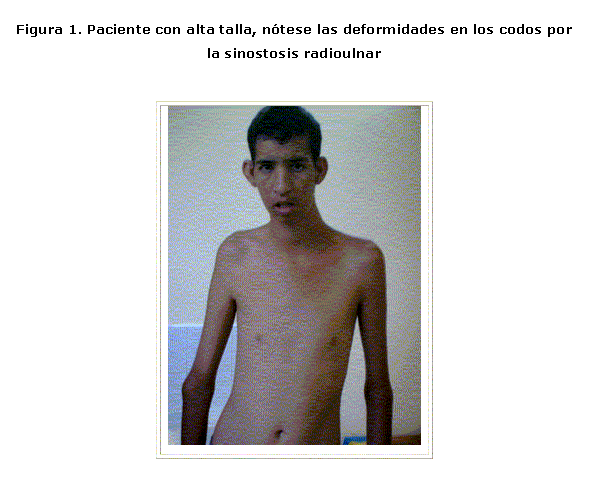
Este paciente no fue escolarizado, ni recibió atención especial, dada su procedencia de un área rural. Figura 1
Para el diagnóstico se aplicó el método clínico a través de la técnica comparativa o de patrón y se confirmó a través del estudio citogenético. Se obtuvo aceptación por parte de los padres para el estudio. Figura 2
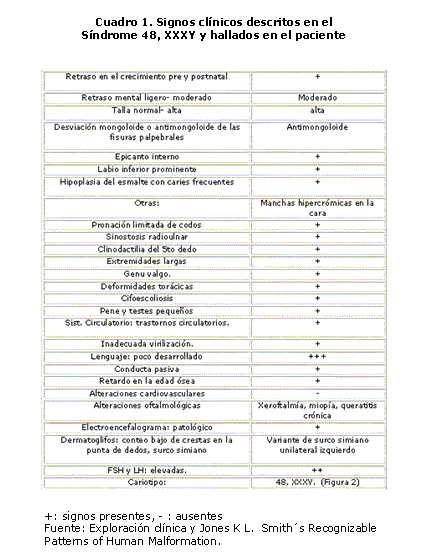
En esta investigación se describen los signos clínicos descritos en el Síndrome 48, XXXY y los que se hallaron en el paciente de este estudio. Cuadro 1
DISCUSIÓN
El síndrome 48, XXXY constituye una variante del síndrome Klinefelter, se presenta en el 5% de éste. En él existen dos cromosomas X supernumerarios, lo que supone un mayor desequilibrio del material genético, siendo proporcional la severidad en el fenotipo y desarrollo cognitivo con el número de cromosomas sexuales extras.3-5 Este fenómeno suele presentarse de forma aislada y se debe a la no disyunción consecutiva en la segregación de los cromosomas X durante la primera y segunda división meiótica ocurridas en la gametogénesis de uno de los padres. El mecanismo por el cual ocurren las polisomías del X permanece incierto, mas, parece ser independiente de la edad materna.6
Con el propósito de determinar el origen parental de los cromosomas extra se realizaron estudios polimórficos de ADN, los cuales se analizaron por técnicas moleculares (reacción en cadena de las polimerasas), donde se comprobó que la mayoría de los casos se aportan por la madre aunque en algunas ocasiones también por el padre, pero siempre el origen ha sido uniparental.3,6,7
Existe gran variabilidad clínica en las aneuploidías de cromosomas sexuales, sin embargo, ésta presenta características propias que las distinguen de las demás y que son evidentes en este paciente: dismorfia facial, sinostosis radioulnar, distribución ginoide y escaso del vello corporal, hipogonadismo hipergonadotrópico, lenguaje poco desarrollado y retraso mental, lo cual coincide con la bibliografía revisada, sin embargo, no se constata la disminución de las crestas dermales en la punta de los dedos, signo importante descrito por Smith.3,8-10
Hay dos aspectos importantes que influyeron de forma negativa en el pronóstico y calidad vida de este paciente: primero, no fue escolarizado, ni recibió enseñanza especial, por tanto no fue estimulado durante sus primeros años de vida, lo cual repercutió sin lugar a dudas en su desarrollo cognitivo. Segundo: no recibió tratamiento sustitutivo hormonal con testosterona, pudiendo aminorar en gran medida los efectos del déficit de esta hormona tan importante para el desarrollo genital, de caracteres sexuales secundarios, óseo y su bienestar de manera general. Todo esto hubiese podido cambiar el curso de sus discapacidades, siendo válido destacar que el poco desarrollo en el lenguaje y el retraso mental del paciente podrían haber sido atenuados si el diagnóstico, la intervención terapéutica y la estimulación psicosensorial hubiesen sido precoces, logrando maximizar sus potencialidades.
Por ello conocer sus principales manifestaciones clínicas son elementos de relevante importancia a tener en cuenta, con fines preventivos.
CONCLUSIONES
El Síndrome 48, XXXY, se caracteriza clínicamente por dismorfia facial, anomalías genitales, esqueléticas y retraso mental de grado variable con afectación en el lenguaje.
El diagnóstico se confirma mediante el estudio citogenético empleando técnicas convencionales (bandas GTG), siendo válido destacar la importancia del diagnóstico precoz para ofrecer oportuno asesoramiento genético a los padres, imponer tratamiento médico y rehabilitar física, psíquica y socialmente al paciente.
REFERENCIAS BIBLIOGRÁFICAS
1. Visootsak J, Graham J M. Klinefelter Syndrome and Its variants [Serie en internet]. [Citado 2004]; [aprox 5 p]. Disponible en: http://www.orpha.net/data/patho/GB/uk-KS.html.
2. Manning MA, Hoyme HE. Diagnóstico y manejo del adolescente con síndrome de Klinefelter. Rev Adolesc Med 2005; 13 (2):27-30. Disponible en: http://www.sap.org.ar/staticfiles/publicaciones/correo/cor3_03/1075.pdf
3. Jones KL. Smith´s Recognizable Patterns of Human Malformation. 5th Ed. Montreal: W.S. Saunders Company; 2004.p.76-7.
4. Catovic A. Phenotype manifestations at polysomy X at males. Bosn J Basic Med Sci 2008;8(3):287-90.
5. Ottesen AM, Garn ID, Aksglaede L, Juul A, Raipert D, Meyts E. A simple screening method for detection of Klinefelter syndrome and other X-chromosome aneuploidies based on copy number of androgen receptor gene. Mol Hum Reprod 2007;13(10):745-50.
6. Leal CA, Belmont JW, Nachtman R, Cantu JM, Medina C. Parental origin of the extra chromosomes in polisomy X. Hum Genet 1994; 94(4):423-6.
7. Lorda-Sanchez I, Binkert F, Hinkel KG, Moser H, Rosenkranz W, Maechler M. Uniparental origin of sex chromosome polysomies. Hum Hered 1992;42(3):193-7.
8. Chen H. Klinefelter syndrome. Disponible en: http://www.emedicine.com/ped/topic1252.htm
9. Visootsak J, Rosner B, Dykens E, Tartaglia N, Graham JM. Behavioral phenotype of sex chromosome aneuploidies: 48,XXYY, 48,XXXY, and 49,XXXXY. Am J Med Genet A 2007;143(11):1198-203.
10. Visootsak J, Graham JM Jr. Klinefelter syndrome and other sex chromosomal aneuploidies. Orphanet J Rare Dis 2006;(1):42.
Recibido: 12 de mayo de 2009
Aprobado: 15 de julio de 2009
Dra. Carmen María Chiong Quesada. Especialista de I Grado en Genética Clínica. Profesor Instructor. Centro Provincial de Genética. Camagüey, Cuba.

{kind=link}
{kind=link}
{kind=link}