










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkBiotecnología Aplicada
versión On-line ISSN 1027-2852
Biotecnol Apl vol.32 no.2 La Habana abr.-jun. 2015
REVIEW
From AZT to treatment as prevention. The evolution of antiretroviral therapy for HIV/AIDS
Del AZT al tratamiento como prevención. La evolución de la terapia antiretroviral contra el VIH/sida
Carlos A Duarte, Taimí Paneque, Anna C Ramírez, Dionne Casillas, Celia Fernández-Ortega
Grupo de Terapia de VIH, Departamento de Farmacéuticos, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.
ABSTRACT
The antiretroviral therapy (ART) for the treatment of HIV/AIDS has been extremely successful in prolonging the lives of people living with HIV. Since the approval of AZT up to the present over 26 individual compounds have been added to the arsenal available for physicians. The combination of three of these drugs, directed against more than one target was the key to achieve a prolonged suppression of viral load with the consequent gain in the life expectancy of the patients. The development of drugs with improved virological and pharmacological properties, less adverse effects and a better resistance profile, together with the application of pharmacological boosters such as ritonavir and cobicistar, the implementation of single pill formulations of drugs to reduce pill burden, and a reduction in the production costs associated to the introduction of generics, have allowed a considerable expansion of the ART coverage in low and middle resource countries. Based on the results from the START clinical trial in 2015, which demonstrated the advantages of the early application of ART in patients with CD4+ T cell counts over 500 cells/mm3, the main regulatory agencies has modified the recommendations about when to start ART. Additionally, the demonstration of the protective effect of ART among discordant couples has open new horizons for the implementation of ART as a preventive intervention. Recently, UNAIDS has launched its new campaign aimed at the expansion of the ART coverage to reach a 90 % reduction in HIV infections by 2030.
Keywords: HIV, AIDS, antiretroviral therapy, prevention.
RESUMEN
La terapia antiretroviral (TAR) ha sido muy exitosa para la prolongación de la vida y la salud de los personas infectadas con el VIH/sida. Desde la aprobación del primero de ellos, el AZT, se han sumado más de 26 compuestos al arsenal terapéutico. La combinación de tres de estos compuestos dirigidos contra más de un blanco fue la clave para lograr una supresión prolongada de la carga viral. El desarrollo reciente de drogas con mejores propiedades antivirales y farmacológicas, efectos adversos menos frecuentes y severos, y un perfil de resistencia más favorable, ha permitido ampliar considerablemente la cobertura entre los pacientes tratados en países de medianos y bajos ingresos, junto a la aplicación de potenciadores farmacológicos como el ritonavir y el cobicistat, la obtención de formulaciones combinadas de drogas para reducir el número de tabletas diarias, así como la disminución de los costos de producción para las variantes genéricas. El ensayo clínico START en 2015 demostró las ventajas de la aplicación inmediata de la TAR en pacientes con conteos de células T CD4+ por encima de 500 células/mm3, y permitió que las agencias reguladoras modificaran las recomendaciones sobre el estadio clínico para el inicio de la TAR. Adicionalmente, la demostración del efecto protector sobre la transmisión del virus en parejas discordantes, abrió un nuevo horizonte para la implementación de la TAR como instrumento preventivo. La meta lanzada por ONUSIDA para ampliar la cobertura terapéutica hasta el 90 % de los pacientes que la necesitan, persigue disminuir hasta en un 90 % las infecciones por VIH para el 2030.
Palabras clave: VIH, sida, terapia antiretroviral, prevención.
INTRODUCTION
Antiretroviral therapy (ART) has been very successful for the treatment of HIV/AIDS. Since the former application in 1996 of the so-called highly active antiretroviral therapy (HAART), HIV/AIDS turned from a fatal into a chronic condition [1-4]. Its treatment efficacy and efficient preventive effect on the virus vertical transmission have been proven [5]. In this review, an overview is presented on the ART therapeutic stages for HIV/AIDS treatment, starting from the early days of the pandemic, when ART was not available and life expectancy was very short since the manifestation of the first symptoms, until current preventive strategies with a better outcome for pandemic eradication.
THE PRE-ART PERIOD
The first cases of an unknown syndrome were reported in US in 1981, characterized by a profound drop in CD4+ T lymphocyte counts and subsequent immune depression of patients. In those days, the disease was called “Gay pest”, “Gay cancer” or Gay-related immune deficiency (GRID), due to its major incidence among men having sex with men (MSM) [6, 7]. The further demonstration that heterosexual patients were equally susceptible to infection led to its official definition as Acquired Immunodeficiency Syndrome (AIDS) [8].
Discovery of the Human Immunodeficiency Syndrome
The race for discovering the etiological agent of AIDS brought about the first results in 1983, when a team directed by the French virologist Luc Montagnier published on the identification of retroviral particles and reverse-transcriptase activity in cultures of lymphocytes isolated from AIDS patients [9]. This was the first report associating a retrovirus with AIDS, but not conclusive on their causal relationship. Less than a year later, the group led by Robert C. Gallo at the National Cancer Institute provided solid evidences in four reports, supporting the hypothesis of a new retrovirus as the causal agent of AIDS [10-13]. The corner stone in Gallo’s work was to replicate the new virus in a tumor cell line of lymphoid origin (H9), providing enough viral material to characterize its proteins and to develop serologic diagnosis methods to detect antibodies specific for the virus in patients’ sera [14]. Consequently, the nucleotide sequences of two different but similar viruses were elucidated, markedly different from any previously identified human retrovirus [15, 16]. This was the basis for denominating the new entity as the Human Immunodeficiency Virus (HIV) [14].
The knowledge of the HIV replicative cycle as the basis for the design of viral inhibitors
Shortly after the discovery of HIV, an intense research effort was conducted in several laboratories worldwide to unravel its viral structure and to characterize the proteins coded in the viral genome. Next, we will summarize the main events of HIV replication cycle. For further details the reader may consult one of the following reviews [17-29].
HIV is an enveloped RNA virus, as every known retrovirus. Its genome encompassing 9.6 kb codes for three types of proteins: structural, enzymatic and regulatory (also called auxiliary).
Structural proteins comprise those located in the viral envelope membrane: gp120 or external glycoprotein, and the gp41 or transmembrane glycoprotein. Other four proteins derived from a common precursor protein of 55 kDa form the viral capsid: the matrix (p17), capsid (p24), nucleocapsid (p9) and p7 proteins
Three proteins display key viral enzyme functions during the viral replication cycle: reverse transcriptase (RT), protease (P) and integrase (I). And five other proteins display regulatory or auxiliary functions, all of them required for an efficient viral replication: Tat [30], Rev [31], Nef [32], Vif [33] and Vpr [11].
The viral particle encapsidates two copies of the RNA viral genome, together with a lysine transfer RNA molecule which functions as primer for the reverse transcription of the viral RNA genome into DNA.
HIV mainly infects CD4+ T lymphocytes, those cells with a key function in the orchestration of the adaptive immune response; also capable of infecting efficiently other cell types such as dendritic cells, macrophages, microglia and Langerhans’ cells, which play an essential role in the immune response [34, 35].
The HIV replicative cycle begins with the recognition of the high affinity viral receptor, the CD4 molecule, on the target cell surface. This first contact induces conformational changes in the gp120, these changes expose a second binding site within the gp120 structure known as the viral co-receptor binding domain. This site attaches to several molecules of the chemokine receptors family, mainly CCR5 and CXCR4 [36-38]. Once completed the interaction of the virus with receptor and co-receptor, another major structural change occurs, releasing the hydrophobic N-terminal domain of the gp41. This domain inserts into the cell membrane, taking both the viral envelope and cell membranes into close proximity [39]. Following the internal retro-traction of the gp41 trimers’ alpha helixes which wrap around each other to put both membranes closer enough, both membranes fuse together, releasing the viral capsid within the cell [40].
Subsequently, the capsid disassembles in the cytoplasm releasing the viral RNA genome, which is copied by the RT to generate a double-stranded DNA molecule to be transported into the nucleus and inserted into the cell DNA by the action of the viral integrase [41, 42]. HIV RT is characterized by its low-fidelity processing during the copy of RNA into DNA. It has been estimated that this enzyme introduces an error every 1000 to 10 000 nucleotides [43-45]. This generates virus particles having at least 1 to 10 mutations per viral genome. Once the viral DNA genome is produced, named provirus and indistinguishable but for its sequence from the human DNA genome, it can be inactive and remain untranscribed for long, undefined periods, with no viral protein production [46-48]. When the infected host cell become activated to certain extent, some transcriptional factors such as NFκB acts on the viral promoter and initiate the transcription of viral genes [49, 51].
During the first early stage of transcription, the process is inefficient and the mRNA molecules are subjected to multiple splicing, only producing the low molecular weight proteins. One of those proteins is Tat, a potent transcriptional transactivator which accumulates into the nucleus and increases over 1000 times the levels of mRNA produced [52]. Another key protein in this process is Rev, also located in the nucleus. It binds to the viral mRNA at a site known as RRE and protects the molecule from multiple splicing, aiding to the formation of single or twice spliced, long transcripts, which reach the cytoplasm and are successfully transcribed to produce the viral RNA proteins and used as viral RNA genome. Then, the capsid proteins assemble to encapsulate two viral genomic RNA molecules within a particle that is directed to the cell membrane and released through a process known as budding. The viral particle becomes enveloped by the cell membrane at the site of budding, coated by the viral glycoprotein spikes previously anchored in the cell membrane at the budding point. Outside the cell, the viral particle undergoes a maturation process, finally structuring the inner shell of the capsid and making the viral particle infective, ready to initiate another infection cycle [53-55]. There has been estimated that a person living with AIDS generates around 109-1010 viral particles daily [56-58].
EVOLUTION OF ART AGAINST HIV/AIDS
In general, the historical evolution of ART against HIV/AIDS can be divided in three discrete periods:
First period (1986-1996): mono- and bitherapy with inhibitors against RT.
Second period (1997-2014): highly active antiretroviral therapy (HAART), also known as tritherapy.
Third period (2015 and beyond): starting by the extension of ART treatment to every person living with HIV, not only for the benefit of patients but also as a weapon to actively prevent viral transmission.
Lets analyze these three periods in further detail.
First period: Failure of mono- and biotherapy with RT inhibitors
ART against HIV started as early as in 1986, after the approval of the first medicine against AIDS by the FDA: azidothymidine or AZT [59]. Between 1981 and 1986, AIDS patients had no medicine available to fight the infection. This caused a dramatic decline in life expectancy to just one year in half the patients developing AIDS, defined as having CD4+ T cell counts below 200 cells/mm3 or characterized by the appearance of well defined opportunistic infections associated to the disease [60].
The first period in the evolution of ART, when one or two combined drugs were applied, did not change much that setting. In spite of attaining certain level of efficacy [61, 62], these drugs did not have a marked impact neither in the intermediate nor the long term of disease progression.
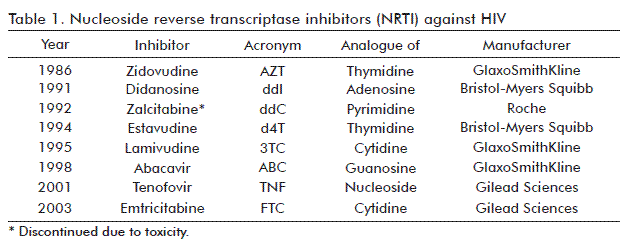
The first viral protein targeted by ART was RT. A series of compounds similar to AZT followed in the years to come, belonging to the family of nucleoside reverse transcriptase inhibitors (NRTI) [63-66] (Table 1). These molecules were called DNA strand terminators, their effects based on the similarity of their structures to some of the nucleotides. RT shows affinity for and erroneously incorporates them into the newly synthesized DNA strand. Nevertheless, due to their lack of an acceptor 3’ hydroxyl group in the deoxyribose molecule, the enzyme is unable to incorporate another nucleotide subsequently, thereby interrupting the DNA strand synthesis.
Remarkably, the weakness of monotherapy was the fast selection for mutants resistant to the drug administered. Almost all these inhibitors can be override by emergent viruses mutated in the RT gene. One of the mechanisms involves the loss of the enzyme capacity to recognize the nucleoside analogue [67, 68]. Alternatively, the mutant enzyme incorporates an ATP-dependent pyrophosphorolytic activity capable of eliminating the NRTI from the 3´ terminus during the elongation of the DNA strand [69-71]. As a result, in just few months, viral load returns to initial levels, this time with resistant viruses.
With the development of nevirapine (NVP; Boheringher Ingelheim) [72. 73], a new type of RT inhibitors appeared: the non-nucleoside RT inhibitors (NNRTI). These compounds tend to be highly hydrophobic, with a structure resembling the wings of a butterfly. They accommodate into the cavity adjacent to the active site of the enzyme, and alosterically inhibit its activity (Figure 3) [74, 75]. Curiously, such a cavity only appears in the presence of the inhibitors. NVP was followed by delavirdine (DLV, ViiV Healthcare) [76, 77]; efavirenz (RPV; Bristol-Myers Squibb) [79] and more recently etravirine (ETR) [80] and rilpavirine (RPV) [81, 82], these last manufactured by Janssen Pharmaceuticals.
The combination of these two types of inhibitors while superior to monotherapy did not significantly improve the clinical outcome of patients, nor steadily reduced viral load [83, 84]. Bitherapy was also a failure for the very same reasons as monotherapy: the emergence of viral resistance as an unbeatable obstacle.
Second period: Implementing HAART was a huge step forward in AIDS therapeutics
HAART
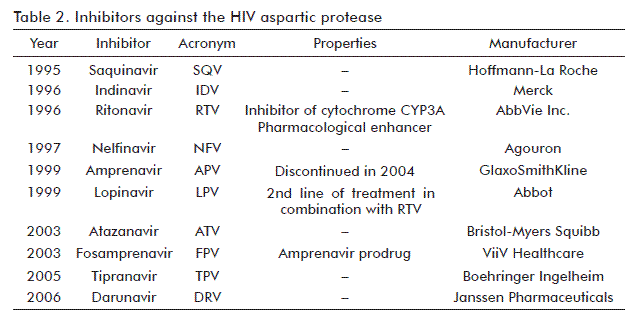
The second evolutionary period of ART was closely related to the development of a third type of drug: protease inhibitors (PI). These small chemical compounds were capable of inserting into the HIV protease active site, further blocking its catalytic activity [85]. The HIV protease belongs to the family of aspartatic proteases [86]. The active form is a homodimer composed by two 11 kDa monomers. Its role in the viral replication cycle is the proteolytic processing of the Gag and Gag-Pol polyproteins to generate the different proteins forming the mature virion [87]. All this proteolytic processing is interrupted by the insertion of PIs into the active site of protease, thereby blocking the formation of mature virions.
The first PI approved by regulatory agencies was saquinavir [88, 89]. Other nine molecules of this type have been subsequently generated [90-93] (Table 2).
At the same time, the use of these inhibitors as monotherapy did not qualitatively fostered ART efficacy. Resistance was also present, although starting with the gradual emergence of viruses carrying primary mutations which impeded the binding of inhibitors to the protease active site, followed by other mutations at least partially compensating the fitness lost with the primary mutations [94].
Hopefully, the results of clinical trials presented at the HIV World Congress held in Vancouver, in 1996, were about to radically transform the ART perspective. The combination of three anti-HIV drugs (two NRTI plus a PI) successfully reduced viral loads down to undetectable levels and kept it under control for more than a year under treatment [95-99].
Two years after implementing HAART, the first data appeared supporting a decrease in mortality and morbidity due to opportunistic infections in treated patients [100, 101]. Although premature, these results revived the optimism not only on the possible control of HIV/AIDS through therapy, but even its eradication. Dr. David Ho at the Diamond Research Center, one of the most renowned virologists at that time on HIV studies, raised the slogan “to hit early and hard” [102]. This necessarily implied to start what was called HAART in asymptomatic patients as early as possible after infection. It was strongly considered the possibility that, if the virus would be silenced for several years, it would be possible to be eradicated itself by the natural immune response of the organism [102, 103]. Unfortunately, such strategy did not stand long enough, due to the abundant and serious adverse effects caused by all these drugs, particularly PIs, in most patients [104-106]. Some of the adverse effects associated to the use of these drugs are listed in table 3.
Therefore, the use of HAART was not properly justified in first instance, due to its cumulative adverse effects and the risk for emergence of resistant viruses, more detrimental than the possible benefits coming from the control of viremia since the very early stages of infection. This led to the decision to postpone its application until the decline of CD4+ T lymphocyte counts below 250 cells/mm3 or until the patient suffering from opportunistic infections defining the AIDS stage. Throughout the years, the tendency has been to increase the number of CD4+ T cells at which such a therapy was started. Today, regulatory agencies emphasize on their recommendations to start HAART at CD4+ T cell counts below 350 cells/mm3, even recommending the evaluation of the limit of 500 cells/mm3 as starting criterion for HAART [107]. These topics will be further addressed.
Other targets for inhibiting HIV replication
T20 and the inhibition of membrane fusion
Biomedical research progressively unraveled the replication mechanisms of HIV and this knowledge brought to light new types of inhibitors. The next success in clinical approval was a membrane fusion inhibitor, therefore blocking viral entry. The compound, known as T20 or enfuvirtide (INN) was developed by Hoffman-La Roche, the first of its kind and the only peptide inhibitor approved to treat HIV/AIDS [108, 109].
T20 is a 36-amino acids peptide extracted from the gp41 protein sequence. It belongs to one of the alpha helix regions that wrap around each other to form a bundle, putting into close contact both membranes. T20 accommodates on its helix counterpart further preventing the formation of the bundle by competition with homologous region within gp41, and ultimately, the fusion of membranes [110].
Nevertheless, enfuvirtide has been limited in the clinical practice due to difficulties in the production scaling up inherent to its peptide nature, its short half life time and its administration by subcutaneous route. Hence, it has been exclusively used as the ultimate line of therapy, when facing the failure of all the treatments available due to the emergence of resistant viruses [111, 112].
Several years after the discovery of T20, another inhibitor capable of blocking viral entry was approved: maraviroc. It was design to interfere the binding of the virus to its co-receptors [113, 114].
Maraviroc as blocker of the HIV binding to the CCR5 co-receptor
Once established the binding mechanisms of the virus to its co-receptors, several pharmaceutical laboratories seek for molecules able to block their interaction. The first of those compounds, maraviroc, was approved in 2007, a small chemical molecule specifically binding to CCR5 and blocking its interaction with gp120 [115]. Maraviroc was developed by Pfizer upon optimization of UK-107,543, an imidazopyridine selected by massive screening through a CCR5-binding assay. More than 1000 analogues were evaluated during the optimization process, in order to improve potency and reduce the compound toxicity [116].
Subsequently, the inhibitor demonstrated to be safe and efficacious in clinical trials, both in patients previously treated with other drugs and in those naïve to treatment [117, 118]. Nevertheless, maraviroc had no commercial success, its weakness residing on its efficacy limited to monocytotropic strains which use CCR5 as co-receptor. This implies that prior to the administration of maraviroc to a patient, it is necessary to investigate on the co-receptor tropism of its resident viral strains [119], an economic cost and a time loss imposed by the complexity of such tests.
Integration inhibitors
The HIV inhibitor is a 32 kDa protein, the major component of the viral integration complex. Its enzyme activity on proviral DNA resides on the elimination of two nucleotides from the 3’ terminus and forming a phosphodiester bong between the terminus and the host chromosomal DNA [120].
In 2007 and after two decades of research, raltegravir (RAL; Merck) appeared as the first integration inhibitor, being approved for clinical application [121]. Two other inhibitors of this type have also been approved so far: dolutegravir (DTG; GlaxoSmithKline) in 2013 [122] and elvitegravir (EVG; Gilead Sciences) in 2014 [123]. Three of them are small molecules for oral administration.
Opposed to the experience with entry inhibitors, raltegravir have had a sound commercial success with significant sales. Such results have being determined by its small size and accessibility, as well as its mechanism of action was not interfered by the emergence of resistant mutant viruses. Moreover, the adverse effects it provokes are milder than those caused by precedent ART drugs (Table 4).
This example illustrates that, in spite of the existence of about 26 drugs available against HIV, the arsenal continuously grows, to fight the continuous emergence of resistant viruses. Even when ART has substantially impacted on the AIDS epidemics, with a radical decrease on mortality and improving the patients’ quality of life [124], the complete eradication of HIV from the body remains a goal to be achieved.
Undoubtedly, ART efficacy has improved over time. Particularly since 2010, the number of therapeutic options available in the clinical setting has increased, reducing viral load down to undetectable levels and progressively lowering it. Additionally, the first choice regimes suppress viral load in more than 90 % of the cases, even after 8 years on therapy [3, 125].
Auxiliary and capsid proteins as targets for ART
A plausible source for targeting HIV is its regulatory proteins, in spite of had been relegated as targets for ART in clinical trials. But, undoubtedly, interfering with their functions would have an immediate impact on HIV replication as experienced with the enzyme viral proteins. For instance, the levels of viral transcription would be reduced as much as 1000 times upon blocking the activity of Tat. In fact, various molecules have been described capable of inhibiting the action of Tat and thereby viral replication [126-129], but just one of them has been evaluated in the clinical setting and unfortunately with negative results [130, 131].
Likewise, neutralizing Rev would limit the synthesis of structural proteins and, therefore, the formation of new virions [132, 133].
In the case of Vif, this protein is essential for virus formation and infectivity, the proof-of-concept of its inhibition validated by testing a small chemical molecule as inhibitory for viral replication [44]. It is highly probable that in forthcoming years some of these strategies would prove efficacious in clinical studies.
Additionally, there are evidences on the possible impact that inhibition of Nef would have on HIV replication, although it is known that Nef-deficient viruses does not lose completely their capacity to replicate, making of this protein a less attractive target [134].
Other interesting targets are the capsid proteins [135]. It has been demonstrated in in vitro experiments that small molecules interfering with the morphogenesis of new virions and their maturation process could inhibit the viral replication [136, 137]. One of these compounds was evaluated in phase I and II clinical trials but its potency was insufficient [138].
New host targets for ART
It is known that HIV receptors use other cellular proteins as auxiliary factors during its replication cycle. Among them, the nuclear proteins Emerin and BAF are involved in the penetration of the integration complex into the nucleus [139-142]. Other proteins take part in the formation of the integration complex and capsid assembly processes. Any of these cellular factors and others remaining to be identified could become an effective target for ART.
Recommended ART treatment regimes
A large number of combinations have been tested with more or less success for antivirals approved for clinical use. Based on cumulative clinical evidences, a panel of experts concerted by the National Institutes of Health of US established the combinations recommended as optimal for clinical use, either as first choice of treatment for patients naïve to treatment or as second line for those whose ART therapy has been unsuccessful or as rescue treatment in patients with changes more than twice in the ART regime [107].
In the following we summarize the updated recommendations of that panel:
First line of treatment for patients starting ART
Combinations using an integrase inhibitors plus 2 NRTIs
• Doluteglavir (DTG)/abacavir (ABC)/lamivudine (3TC).
• Doluteglavir DTG /tenofovir (TDF) /emtricitabine (FTC) (only for patients negative for HLA-B*5701).
• Elvitegravir (EVG)/cobicistat (c)/ tenofovir (TDF) / emtricitabine (FTC).
• Raltegravir (RAL)/ tenofovir (TDF) /emtricitabine (FTC).
Combinations including protease inhibitors (PI)
• Darunavir (DRV)/ritonavir (r) / tenofovir (TDF) /emtricitabine (FTC).
Cobicistat and ritonavir are used in these regimes as enhancers of other inhibitors’ effects. These two compounds inhibit liver enzymes belonging to the subfamily of Cytochrome P450 3A4, which metabolize other inhibitors and thereby increase their pharmacokinetic properties, supporting a possible reduction of the dosage and the number of applications [143]. They differ in that ritonavir is simultaneously an HIV PI although ineffective at the concentrations administered in these regimes, while cobicistat is a pharmacokinetic booster with no direct action on HIV replication.
There are a series of alternative regimes recommended solely for those situations in which the previously mentioned combinations cannot be used. All of them are triple combinations, except for the lopinavir (LPV)/r / lamivudine (3TC) and darunavir (DRV)/r /raltegravir (RAL) dual regimes, only recommended for patients intolerant to tenofovir and abacavir.
Cases of virologic failure. Second line of ART treatment
Virologic failure is defined as the impossibility for maintaining viral RNA levels below 200 copies/mm3 under ART. The causes are varied and its analysis is beyond the scope of this review. But it is worth to mention that, in those cases, there have to be checked if deficient adherence to treatment occurred together with the characterization of the resistance pattern of the viruses infecting the patient. Depending on the results, another ART regime must be proposed, with drugs effective against the virus variants present, the drugs either aimed towards a different target or able to neutralize the viruses resistant to drugs of the same type.
Repeated virologic failure
Considering the current drug diversity, the number of people presenting virologic failure against two or more ART regimes has decreased. Nevertheless, such hard to treat cases could occur, commonly associated to viral strains showing resistance against various types of inhibitors. Therefore, the inclusion of inhibitors very different from those recommended, such as T20 or maraviroc, could be very helpful. Ultimately, if the viral load could not be completely controlled below the detection limits of available tests, ART could be applied to maintain a partial control on viral replication levels. This will always provide a more favorable prognosis for patients than leaving them with no therapy.
The rise back of bitherapy?
There are renewed attempts to reduce to just two the number of inhibitors included in ART regimes, fueled up by the availability of integrase inhibitors, second and third generations’ drugs against RT and protease, with improved potency, a more favorable viral resistance profile and less adverse effects than previous drugs. In this sense, various clinical studies have demonstrated that biotherapy has similar effects than the recommended tritherapy.
The first of such studies, denominated GARDEL (Global AntiRetroviral Design Encompassing Lopinavir/r and lamivudine vs LPV/r based standard therapy), demonstrated that the lopinavir (LPR)/r /lamivudine combination was not inferior to the triple combinations of lopinavir (LPR)/r /lamivudine (3TC) or emtricitabine (FTC)/other NRTI [144]. It is worth to mention that, in this trial, the effect was measured only for 48 weeks, that is, less than a year of treatment and the regimes used for comparing biotherapy were alternative, second line regimes, not those recommended as optimal by the experts.
A second trial, named PROGRESS, was a pilot, randomized, open trial, run to compare safety and efficacy after 96 weeks of a regime administering lopinavir (LPV)/r/raltegravir (RAL) twice-a-day against the regime with three inhibitors Tenofovir (TDF)/emcitrabine (FTC)/ LPV/r once-a-day. The results indicated that bitherapy was not inferior, this time for a longer period, but the limited proportion of patients having a viral load of more than 100 000 copies at the start of the study made difficult to assess the efficacy of this strategy [145].
In the third study, NEAT001/ANRS143, the efficacy was evaluated for a regime based on darunavir (DRV)/ r/raltegravir (RAL) administered twice a day for 96 weeks as compared to a triple regime administering tenofovir (TNF)/emtricitabine (FTC)/darunavir (DRV)/r. This trial concluded with no superiority of the standard treatment over bitherapy to control viral load [146].
These results show a favorable tendency supporting the long term evaluation of such optimized bitherapy regimes, aimed to reduce ART costs and their associated adverse effects. Nevertheless, more clinical testing is required to validate the long term effect of these formulations prior to recommending them as first line of treatment for patients naïve to ART.
Combo administration: strategies to reduce the number of pills
One of the difficulties to achieve the adequate adherence of patients to ART is the high pill burden they have to take daily. To solve this problem, the industry has developed combined formulations which integrate more than one inhibitor in the same pill. Some of those formulations are:
• Atripla®, the first once-a-day pill, a turning point for ART combined administration. It was developed by Bristol-Myers Squibb and contains efavirenz (EFV) with tenofovir (TFV) and emtricitabine (FTC). This combination, nevertheless, is not among the recommended ones to initiate ART.
• Complera®, manufactured by GILEAD, combines rilpivirin (RPV) with tenofovir (TFV) and emtricitabine (FTC) and is recommended for patients with viral loads below 100 000 copies/mL. It is not among the regimes recommended as first line of treatment.
• Triumeq®, combining dolutegravir (DTV) plus abacavir (ABC) and lamivudin (3TC), is manufactured by ViiV Healthcare [147].
In the same line, two other formulations of three ART drugs and a pharmacokinetic boosting agent (with no action on HIV: cobicistat) are available from Gilead Sciences, both as single pills: Stribild® and Genvoya®. Each combines an integrase inhibitor (efavirenz or elvitegravir) with two other inhibitors (emtricitabine/tenofovir) [148]. Moreover, Stribild® uses 300 mg of tenofovir fumarate, while Genvoya® uses just 10 mg of tenofovir alafenamide which is more specific for the target and induces less frequent adverse events.
There are also tens of combinations of two inhibitors available, but the predominant tendency is to achieve the highest simplification of treatments by administering a single pill a day due to the positive impact this has on the adherence of patients to ART treatment.
The Test and Treat strategy or back to start
The improvement of ART treatment in terms of efficacy (i.e., the number of options available to fight viral resistance or more tolerable new inhibitors) has led to a reappraisal on when would be appropriate to start treatment.
On this aspect, a panel of experts from the NIH recommended in 2014 to administer ART to all the HIV+ patients to reduce the risks of progression to AIDS [107].
The strength and evidence of this recommendation changes attending to CD4+ T cell counts:
- Below 350 cells/mm3: Strong, validated in randomized control trials;
- Between 350 and 500 cells/mm3: Strong, based on cohort studies or non-randomized studies;
- Above 500 cells/mm3: Moderate, based on experts’ opinions.
With the aim to objectively settle down the debate on the possible benefit for patients to start ART with CD4+ T cell counts above 500 cells/mm3, a study coded START was conducted from 2009 to 2015, enrolling 1500 patients fulfilling such criteria. Half the patients started ART immediately, and the other half when reaching 350 cells/mm3 as recommended by current guidelines. An unquestionable benefit was demonstrated in patients starting therapy with counts above 500 cells/mm3 in terms of preventing or delaying the progression to AIDS, decreasing the incidence of other severe diseases not associated to AIDS and mortality [149].
Third period: 2015 and beyond
A new age in ART treatment against HIV is about to begin, transcending the ART therapeutic function and providing it a preventive and key role in pandemic control.
For years, ART has been strongly recommended to seropositive mothers to prevent the mother to child infection during fetal development [150]. Its usefulness has also been demonstrated to prevent infection in the case of accidental exposure to the virus and in the prophylaxis prior to getting into contact with it [151].
It has been repeatedly proposed that the expansion of ART among seropositive patients could significantly impact on controlling viral transmission [1]. Mathematical modeling of data from populations treated with ART for long periods supports such possibility [125].
On the other hand, ART has proven efficacious to reduce the risk of transmission either by intravenous route [153] or heterosexual contact [154].
A clinical trial named HPTN 052, initiated in 2005 and concluded in 2011 unequivocally demonstrated the preventive potentialities of ART [5]. It enrolled 1763 discordant sexual couples in 13 sites of 9 countries from Africa, Asia and the US. All the seropositive patients showed CD4+ T cell counts in the range 350-500 cells/mm3. A first group started receiving ART when reaching 250 cells/mm3 or the occurrence of an opportunistic infection, as indicated by regulations enforced at that time. In this group, 27 new infections were demonstrated among the 877 couples.
In the second group, ART was immediately administered. At the end of the study, just one out of the 877 seropositive individuals on this group became infected. These numbers indicated that the treatment with ART in patients with T CD4+ cell counts was 96.6 % effective in the prevention of HIV transmission among sexual couples.
These results together with condom use are the best preventive option and eradication strategy available to control the expansion of the pandemic. In fact, they are by far superior to the 50 % of efficacy demonstrated for male circumcision and the 31.2 % reported for the vaccine candidate combining the Canarypox vector from Pasteur Merieux and the gp120 formulated in Alum from Merck [155]. This last results were at the very limit of statistical significance and insufficient to support its further administration as experimental vaccine.
Additionally, UNAIDS has lunched the 90 × 90 × 90 campaign to expand ART to all seropositive people, with the slogan Test and Treat [156]. Its main purpose is to make the treatment available to the more than 11.7 million of people infected in low and medium income countries (according to reports in 2013), until reaching the 28 million of people eligible for ART attending to their respective national regulations. In this scenario, a probable, long term control of the epidemic is proposed by mathematical models [157]. Results expected from this campaign look too much optimistic, with a 90 % reduction in HIV infections been predicted for 2030, also with a 90 % reduction in AIDS deaths.
CURRENT LIMITATIONS OF ART
Despite the unquestionable success of ART, significant aspects remain to be solved. Let’s take a look at current limitations:
ART does not eliminate completely HIV [124]. Viral persistence relates to the silent infection of long live cells that the virus uses as reservoirs, such as memory T cells [158-161]. There are other organs regarded as “immunological sanctuaries”, such as testicles and the central nervous system, where only a very weak immune response can be entangled. All these makes of ART a treatment for life [162, 163].
Significant adverse reactions persist in patients under treatment, in spite of the new generation ARTs being tolerated better. Decreased adherence of patients to treatment, at least temporarily, is the direct consequence of their associated adverse effects, with viral replication levels increasing as consequence together with the probability of the selection of new mutants that could emerge over time [164].
Viral resistance to ART is a long term issue. Although of delayed appearance, multiresistant viruses finally emerge after several years under treatment, further requiring a change in the therapeutic combination [164, 165].
The high costs of ART are still a problem for developing and underdeveloped countries. It is worth to mention the significant reduction in the costs of ART derived from the increasing market of generics [166], particularly in India [167], which has markedly lowered the costs for low income countries, in addition to the patent expiry of earlier drugs. Nevertheless, the most recent drugs still are economically unaffordable, such as integrase inhibitors and the last generation of NRTIs. The UNAIDS Program has developed a massive campaign to provide therapeutic coverage of the most deficient regions. According to recent reports, the therapeutic coverage in Africa reached up to 37 % among the people living with HIV [168], this resulting from such international efforts in coordination with national programs.
For all these reasons, new ARTs are required, aiming at new therapeutic targets and active against strains resistant to the existing drugs, with improved toxicity profiles, better penetration into the so-called immunological sanctuaries and at lower costs.
CONCLUSIONS
As extensively discussed herein, the goal of turning HIV infection into a chronic treatable disease has been achieved thanks to the tremendous improvements in ART. Such therapeutic means have been achieved after serious research and a long way of trial-and-error still to go, together with sound advances in the comprehension of basic human immunology, virus-host interactions and an extensive insight into the molecular biology of the HIV virus. This has required a huge amount of economic resources and the concerted will to coordinate efforts enough in achieving such tremendous goals, only rivaled by those successfully eradicating smallpox in the XX century. Hence, when facing current difficulties to obtain a preventive vaccine, ART seems to be the most promising choice offhand to control or even eradicate the AIDS pandemic.
REFERENCES
1. Arts EJ, Hazuda DJ. HIV-1 antiretroviral drug therapy. Cold Spring Harb Perspect Med. 2012;2(4):a007161.
2. Fauci AS, Folkers GK, Dieffenbach CW. HIV-AIDS: much accomplished, much to do. Nat Immunol. 2013;14(11):1104-7.
3. Sabin CA. Do people with HIV infection have a normal life expectancy in the era of combination antiretroviral therapy? BMC Med. 2013;11:251.
4. Deeks SG, Lewin SR, Havlir DV. The end of AIDS: HIV infection as a chronic disease. Lancet. 2013;382(9903):1525-33.
5. Cohen MS, Chen YQ, McCauley M, Gamble T, Hosseinipour MC, Kumarasamy N, et al. Prevention of HIV-1 infection with early antiretroviral therapy. N Engl J Med. 2011;365(6):493-505.
6. Centers for Disease C. A cluster of Kaposi’s sarcoma and Pneumocystis carinii pneumonia among homosexual male residents of Los Angeles and Orange Counties, California. MMWR Morb Mortal Wkly Rep. 1982;31(23):305-7.
7. Gottlieb MS. Pneumocystis pneumonia--Los Angeles. 1981. Am J Public Health. 2006;96(6):980-1.
8. Quagliarello V. The Acquired Immunodeficiency Syndrome: current status. Yale J Biol Med. 1982;55(5-6):443-52.
9. Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science. 1983;220(4599):868-71.
10. Schupbach J, Popovic M, Gilden RV, Gonda MA, Sarngadharan MG, Gallo RC. Serological analysis of a subgroup of human T-lymphotropic retroviruses (HTLV-III) associated with AIDS. Science. 1984;224(4648):503-5.
11. Popovic M, Sarngadharan MG, Read E, Gallo RC. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre- AIDS. Science. 1984;224(4648):497-500.
12. Sarngadharan MG, Popovic M, Bruch L, Schupbach J, Gallo RC. Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science. 1984;224(4648):506-8.
13. Gallo RC, Salahuddin SZ, Popovic M, Shearer GM, Kaplan M, Haynes BF, et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science. 1984;224(4648):500-3.
14. Weiss SH, Goedert JJ, Sarngadharan MG, Bodner AJ, Group ASCW, Gallo RC, et al. Screening test for HTLV-III (AIDS agent) antibodies: specificity, sensitivity, and applications. JAMA. 2014;312(4):442.
15. Wain-Hobson S, Sonigo P, Danos O, Cole S, Alizon M. Nucleotide sequence of the AIDS virus, LAV. Cell. 1985;40(1):9-17.
16. Ratner L, Fisher A, Jagodzinski LL, Mitsuya H, Liou RS, Gallo RC, et al. Complete nucleotide sequences of functional clones of the AIDS virus. AIDS Res Hum Retroviruses. 1987;3(1):57-69.
17. Wong-Staal F, Ratner L, Shaw G, Hahn B, Harper M, Franchini G, et al. Molecular biology of human T-lymphotropic retroviruses. Cancer Res. 1985;45(9 Suppl):4539s-44s.
18. Bryant ML, Ratner L. Biology and molecular biology of human immunodeficiency virus. Pediatr Infect Dis J. 1992;11(5):390-400.
19. Harada S, Ratner L. AIDS 1997. Virology: overview. AIDS. 1997;11 Suppl A:S1-2.
20. Jeang KT. HIV-1: molecular biology and pathogenesis. Adv Pharmacol. 2000;48:xvii-xix.
21. Freed EO. HIV-1 replication. Somat Cell Mol Genet. 2001;26(1-6):13-33.
22. Ferguson MR, Rojo DR, von Lindern JJ, O’Brien WA. HIV-1 replication cycle. Clin Lab Med. 2002;22(3):611-35.
23. Tozser J. Stages of HIV replication and targets for therapeutic intervention. Curr Top Med Chem. 2003;3(13):1447-57.
24. Freed EO, Mouland AJ. The cell biology of HIV-1 and other retroviruses. Retrovirology. 2006;3:77.
25. Alcami J. The HIV replication cycle. Established therapeutic targets and potential targets. Enferm Infecc Microbiol Clin. 2008;26 Suppl 12:3-10.
26. Das AT, Berkhout B. HIV-1 evolution: frustrating therapies, but disclosing molecular mechanisms. Philos Trans R Soc Lond B Biol Sci. 2010;365(1548):1965-73.
27. Bieniasz PD. An overview of intracellular interactions between immunodeficiency viruses and their hosts. AIDS. 2012;26(10):1243-54.
28. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8):a006866.
29. Freed EO. HIV-1 assembly, release and maturation. Nat Rev Microbiol. 2015;13(8):484-96.
30. Pugliese A, Vidotto V, Beltramo T, Petrini S, Torre D. A review of HIV-1 Tat protein biological effects. Cell Biochem Funct. 2005;23(4):223-7.
31. Pollard VW, Malim MH. The HIV-1 Rev protein. Annu Rev Microbiol. 1998;52:491-532.
32. Bandres JC, Shaw AS, Ratner L. HIV-1 Nef protein downregulation of CD4 surface expression: relevance of the lck binding domain of CD4. Virology. 1995;207(1):338-41.
33. Sheehy AM, Gaddis NC, Choi JD, Malim MH. Isolation of a human gene that inhibits HIV-1 infection and is suppressed by the viral Vif protein. Nature. 2002;418(6898):646-50.
34. Dalgleish AG, Beverley PC, Clapham PR, Crawford DH, Greaves MF, Weiss RA. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312(5996):763-7.
35. Klatzmann D, Champagne E, Chamaret S, Gruest J, Guetard D, Hercend T, et al. T-lymphocyte T4 molecule behaves as the receptor for human retrovirus LAV. Nature. 1984;312(5996):767-8.
36. Doranz BJ, Rucker J, Yi Y, Smyth RJ, Samson M, Peiper SC, et al. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR- 3, and CKR-2b as fusion cofactors. Cell. 1996;85(7):1149-58.
37. Jentsch S. When proteins receive deadly messages at birth. Science. 1996;271(5251):955-6.
38. Alkhatib G, Combadiere C, Broder CC, Feng Y, Kennedy PE, Murphy PM, et al. CC CKR5: a RANTES, MIP-1alpha, MIP-1beta receptor as a fusion cofactor for macrophage-tropic HIV-1. Science. 1996;272(5270):1955-8.
39. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381(6584):661-6.
40. Cai L, Gochin M, Liu K. Biochemistry and biophysics of HIV-1 gp41 - membrane interactions and implications for HIV-1 envelope protein mediated viral-cell fusion and fusion inhibitor design. Curr Top Med Chem. 2011;11(24):2959-84.
41. Wilen CB, Tilton JC, Doms RW. HIV: cell binding and entry. Cold Spring Harb Perspect Med. 2012;2(8). DOI: 10.1101/cshperspect.a006866.
42. Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009;276(11):2926-46.
43. Craigie R, Bushman FD. HIV DNA integration. Cold Spring Harb Perspect Med. 2012;2(7):a006890.
44. Mansky LM, Temin HM. Lower in vivo mutation rate of human immunodeficiency virus type 1 than that predicted from the fidelity of purified reverse transcriptase. J Virol. 1995;69(8):5087-94.
45. O’Neil PK, Sun G, Yu H, Ron Y, Dougherty JP, Preston BD. Mutational analysis of HIV-1 long terminal repeats to explore the relative contribution of reverse transcriptase and RNA polymerase II to viral mutagenesis. J Biol Chem. 2002;277(41):38053-61.
46. Abram ME, Ferris AL, Shao W, Alvord WG, Hughes SH. Nature, position, and frequency of mutations made in a single cycle of HIV-1 replication. J Virol. 2010;84(19):9864-78.
47. Battistini A, Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses. 2014;6(4):1715-58.
48. Van Lint C, Bouchat S, Marcello A. HIV-1 transcription and latency: an update. Retrovirology. 2013;10:67.
49. Liu RD, Wu J, Shao R, Xue YH. Mechanism and factors that control HIV-1 transcription and latency activation. J Zhejiang Univ Sci B. 2014;15(5):455-65.
50. Taube R, Peterlin M. Lost in transcription: molecular mechanisms that control HIV latency. Viruses. 2013;5(3):902-27.
51. Roulston A, Lin R, Beauparlant P, Wainberg MA, Hiscott J. Regulation of human immunodeficiency virus type 1 and cytokine gene expression in myeloid cells by NF-kappa B/Rel transcription factors. Microbiol Rev. 1995;59(3):481-505.
52. Karn J, Stoltzfus CM. Transcriptional and posttranscriptional regulation of HIV-1 gene expression. Cold Spring Harb Perspect Med. 2012;2(2):a006916.
53. Sundquist WI, Krausslich HG. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2(7):a006924.
54. Briggs JA, Riches JD, Glass B, Bartonova V, Zanetti G, Krausslich HG. Structure and assembly of immature HIV. Proc Natl Acad Sci U S A. 2009;106(27):11090-5.
55. Bukrinskaya AG. HIV-1 assembly and maturation. Arch Virol. 2004;149(6):1067-82.
56. Ho DD, Neumann AU, Perelson AS, Chen W, Leonard JM, Markowitz M. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373(6510):123-6.
57. Wei X, Ghosh SK, Taylor ME, Johnson VA, Emini EA, Deutsch P, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373(6510):117-22.
58. Perelson AS, Neumann AU, Markowitz M, Leonard JM, Ho DD. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271(5255):1582-6.
59. Mitsuya H, Weinhold KJ, Furman PA, St Clair MH, Lehrman SN, Gallo RC, et al. 3’-Azido-3’-deoxythymidine (BW A509U): an antiviral agent that inhibits the infectivity and cytopathic effect of human T-lymphotropic virus type III/lymphadenopathy-associated virus in vitro. Proc Natl Acad Sci USA. 1985;82(20):7096-100.
60. Rothenberg R, Woelfel M, Stoneburner R, Milberg J, Parker R, Truman B. Survival with the acquired immunodeficiency syndrome. Experience with 5833 cases in New York City. N Engl J Med. 1987;317(21):1297-302.
61. Fischl MA, Richman DD, Grieco MH, Gottlieb MS, Volberding PA, Laskin OL, et al. The efficacy of azidothymidine (AZT) in the treatment of patients with AIDS and AIDS-related complex. A double-blind, placebo-controlled trial. N Engl J Med. 1987;317(4):185-91.
62. Buira E, Gatell JM, Miro JM, Batalla J, Zamora L, Mallolas J, et al. Influence of treatment with zidovudine (ZDV) on the long-term survival of AIDS patients. J Acquir Immune Defic Syndr. 1992;5(7):737-42.
63. Furman PA, Fyfe JA, St Clair MH, Weinhold K, Rideout JL, Freeman GA, et al. Phosphorylation of 3’-azido-3’-deoxythymidine and selective interaction of the 5’-triphosphate with human immunodeficiency virus reverse transcriptase. Proc Natl Acad Sci U S A. 1986;83(21):8333-7.
64. Balzarini J, Herdewijn P, De Clercq E. Differential patterns of intracellular metabolism of 2’,3’-didehydro-2’,3’-dideoxythymidine and 3’-azido-2’,3’-dide¬oxythymidine, two potent anti-human immunodeficiency virus compounds. J Biol Chem. 1989;264(11):6127-33.
65. Hart GJ, Orr DC, Penn CR, Figueiredo HT, Gray NM, Boehme RE, et al. Effects of (-)-2’-deoxy-3’-thiacytidine (3TC) 5’-triphosphate on human immunodeficiency virus reverse transcriptase and mammalian DNA polymerases alpha, beta, and gamma. Antimicrob Agents Chemother. 1992;36(8):1688-94.
66. Richman DD. HIV chemotherapy. Nature. 2001;410(6831):995-1001.
67. Schinazi RF, Lloyd RM, Jr., Nguyen MH, Cannon DL, McMillan A, Ilksoy N, et al. Characterization of human immunodeficiency viruses resistant to oxathiolane-cytosine nucleosides. Antimicrob Agents Chemother. 1993;37(4):875-81.
68. Quan Y, Gu Z, Li X, Li Z, Morrow CD, Wainberg MA. Endogenous reverse transcription assays reveal high-level resistance to the triphosphate of (-)2’-dideoxy-3’-thiacytidine by mutated M184V human immunodeficiency virus type 1. J Virol. 1996;70(8):5642-5.
69. Arion D, Kaushik N, McCormick S, Borkow G, Parniak MA. Phenotypic mechanism of HIV-1 resistance to 3’-azido-3’-deoxythymidine (AZT): increased polymerization processivity and enhanced sensitivity to pyrophosphate of the mutant viral reverse transcriptase. Biochemistry. 1998;37(45):15908-17.
70. Meyer PR, Matsuura SE, Mian AM, So AG, Scott WA. A mechanism of AZT resis¬tance: an increase in nucleotide-dependent primer unblocking by mutant HIV-1 reverse transcriptase. Mol Cell. 1999;4(1):35-43.
71. Naeger LK, Margot NA, Miller MD. ATP-dependent removal of nucleoside reverse transcriptase inhibitors by human immunodeficiency virus type 1 reverse transcriptase. Antimicrob Agents Chemother. 2002;46(7):2179-84.
72. Bardsley-Elliot A, Perry CM. Nevirapine: a review of its use in the prevention and treatment of paediatric HIV infection. Paediatr Drugs. 2000;2(5):373-407.
73. Milinkovic A, Martinez E. Nevirapine in the treatment of HIV. Expert Rev Anti Infect Ther. 2004;2(3):367-73.
74. Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;256(5065):1783-90.
75. Sluis-Cremer N, Temiz NA, Bahar I. Conformational changes in HIV-1 reverse transcriptase induced by nonnucleoside reverse transcriptase inhibitor binding. Curr HIV Res. 2004;2(4):323-32.
76. Freimuth WW. Delavirdine mesylate, a potent non-nucleoside HIV-1 reverse transcriptase inhibitor. Adv Exp Med Biol. 1996;394:279-89.
77. Scott LJ, Perry CM. Delavirdine: a review of its use in HIV infection. Drugs. 2000;60(6):1411-44.
78. Maggiolo F. Efavirenz: a decade of clinical experience in the treatment of HIV. J Antimicrob Chemother. 2009;64(5):910-28.
79. Plosker GL, Perry CM, Goa KL. Efavirenz: a pharmacoeconomic review of its use in HIV infection. Pharmacoeconomics. 2001;19(4):421-36.
80. Minuto JJ, Haubrich RH. Etravirina A second generation NNRTI for treatment-experienced adults with resistant HIV-1 infection. Future HIV Ther. 2008;2:525-37.
81. Putcharoen O, Kerr SJ, Ruxrungtham K. An update on clinical utility of rilpivirine in the management of HIV infection in treatment-naive patients. HIV AIDS (Auckl). 2013;5:231-41.
82. Ripamonti D, Bombana E, Rizzi M. Rilpivirine: drug profile of a second-generation non-nucleoside reverse transcriptase HIV-inhibitor. Expert Rev Anti Infect Ther. 2014;12(1):13-29.
83. Eron JJ, Benoit SL, Jemsek J, MacArthur RD, Santana J, Quinn JB, et al. Treatment with lamivudine, zidovudine, or both in HIV-positive patients with 200 to 500 CD4+ cells per cubic millimeter. North American HIV Working Party. N Engl J Med. 1995;333(25):1662-9.
84. Hammer SM, Katzenstein DA, Hughes MD, Gundacker H, Schooley RT, Haubrich RH, et al. A trial comparing nucleoside monotherapy with combination therapy in HIV-infected adults with CD4 cell counts from 200 to 500 per cubic millimeter. AIDS Clinical Trials Group Study 175 Study Team. N Engl J Med. 1996;335(15):1081-90.
85. Valdez H, Lederman MM, Woolley I, Walker CJ, Vernon LT, Hise A, et al. Human immunodeficiency virus 1 protease inhibitors in clinical practice: predictors of virological outcome. Arch Intern Med. 1999;159(15):1771-6.
86. Seelmeier S, Schmidt H, Turk V, von der Helm K. Human immunodeficiency virus has an aspartic-type protease that can be inhibited by pepstatin A. Proc Natl Acad Sci U S A. 1988;85(18):6612-6.
87. Park J, Morrow CD. Mutations in the protease gene of human immunodeficiency virus type 1 affect release and stability of virus particles. Virology. 1993;194(2):843-50.
88. Bragman K. Saquinavir: an HIV proteinase inhibitor. Adv Exp Med Biol. 1996;394:305-17.
89. Noble S, Faulds D. Saquinavir. A review of its pharmacology and clinical potential in the management of HIV infection. Drugs. 1996;52(1):93-112.
90. Andreoni M, Perno CF. Positioning of HIV-protease inhibitors in clinical practice. Eur Rev Med Pharmacol Sci. 2012;16(1):10-8.
91. Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl). 2015;7:95-104.
92. Tejerina F, Bernaldo de Quiros JC. Protease inhibitors as preferred initial regimen for antiretroviral-naive HIV patients. AIDS Rev. 2011;13(4):227-33.
93. Zha W, Zha BS, Zhou F, Zhou H, Wang G. The cellular pharmacokinetics of HIV protease inhibitors: current knowledge and future perspectives. Curr Drug Metab. 2012;13(8):1174-83.
94. Molla A, Korneyeva M, Gao Q, Vasavanonda S, Schipper PJ, Mo HM, et al. Ordered accumulation of mutations in HIV protease confers resistance to ritonavir. Nat Med. 1996;2(7):760-6.
95. Collier AC, Coombs RW, Schoenfeld DA, Bassett RL, Timpone J, Baruch A, et al. Treatment of human immunodeficiency virus infection with saquinavir, zidovudine, and zalcitabine. AIDS Clinical Trials Group. N Engl J Med. 1996;334(16):1011-7.
96. D’Aquila RT, Hughes MD, Johnson VA, Fischl MA, Sommadossi JP, Liou SH, et al. Nevirapine, zidovudine, and didanosine compared with zidovudine and didanosine in patients with HIV-1 infection. A randomized, double-blind, placebo-controlled trial. National Institute of Allergy and Infectious Diseases AIDS Clinical Trials Group Protocol 241 Investigators. Ann Intern Med. 1996;124(12):1019-30.
97. Staszewski S, Miller V, Rehmet S, Stark T, De Cree J, De Brabander M, et al. Virological and immunological analysis of a triple combination pilot study with loviride, lamivudine and zidovudine in HIV-1-infected patients. AIDS. 1996;10(5):F1-7.
98. Gulick RM, Mellors JW, Havlir D, Eron JJ, Gonzalez C, McMahon D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N Engl J Med. 1997;337(11):734-9.
99. Hammer SM, Squires KE, Hughes MD, Grimes JM, Demeter LM, Currier JS, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N Engl J Med. 1997;337(11):725-33.
100. Palella FJ, Delaney KM, Moorman AC, Loveless MO, Fuhrer J, Satten GA, et al. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. HIV Outpatient Study Investigators. N Engl J Med. 1998;338(13):853-60.
101. Jahnke N, Montessori V, Hogg R, Anis AH, O’Shaughnessy M, Montaner JSG. Impact of Triple Drug Therapy on Morbidity Mortality and Cost. AIDS Rev. 1999;1: 57-60.
102. Ho DD. Time to hit HIV, early and hard. N Engl J Med. 1995;333(7):450-1.
103. Bryan CS. Hit early, hit hard: new strategies for HIV. J S C Med Assoc. 1996;92(8):361-3.
104. Harrington M, Carpenter CC. Hit HIV-1 hard, but only when necessary. Lancet. 2000; 355(9221):2147-52.
105. Rodriguez E. Early treatment may not be best. Posit Living. 1999;8:46.
106. Simmons P. When paradigms fall: the demise of “hit hard, hit early”. Res Initiat Treat Action. 1999;5(5):11-2.
107. Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the use of antiretroviral agents in HIV-1-infected adults and adolescents. Bethesda: Department of Health and Human Services, NIH. 2015 [cited 2015 Apr 14]. Available from: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf
108. Kilby JM, Hopkins S, Venetta TM, DiMassimo B, Cloud GA, Lee JY, et al. Potent suppression of HIV-1 replication in humans by T-20, a peptide inhibitor of gp41-mediated virus entry. Nat Med. 1998;4(11): 1302-7.
109. Lalezari JP, Henry K, O’Hearn M, Montaner JS, Piliero PJ, Trottier B, et al. Enfuvirtide, an HIV-1 fusion inhibitor, for drug-resistant HIV infection in North and South America. N Engl J Med. 2003;348(22):2175-85.
110. Eckert DM, Kim PS. Design of potent inhibitors of HIV-1 entry from the gp41 N-peptide region. Proc Natl Acad Sci USA. 2001;98(20):11187-92.
111. Introcaso CE, Hines JM, Kovarik CL. Cutaneous toxicities of antiretroviral therapy for HIV: part II. Nonnucleoside reverse transcriptase inhibitors, entry and fusion inhibitors, integrase inhibitors, and immune reconstitution syndrome. J Am Acad Dermatol. 2010;63(4):563-9; quiz 9-70.
112. Manfredi R, Sabbatani S. A novel antiretroviral class (fusion inhibitors) in the management of HIV infection. Present features and future perspectives of enfuvirtide (T-20). Curr Med Chem. 2006;13(20):2369-84.
113. Van Der Ryst E. Maraviroc - A CCR5 Antagonist for the Treatment of HIV-1 Infection. Front Immunol. 2015;6:277.
114. van Lelyveld SF, Wensing AM, Hoepelman AI. The MOTIVATE trials: maraviroc therapy in antiretroviral treatment-experienced HIV-1-infected patients. Expert Rev Anti Infect Ther. 2012;10(11):1241-7.
115. FDA approves maraviroc tablets. AIDS Patient Care STDS. 2007;21(9):702.
116. Dorr P, Westby M, Dobbs S, Griffin P, Irvine B, Macartney M, et al. Maraviroc (UK-427,857), a potent, orally bioavailable, and selective small-molecule inhibitor of chemokine receptor CCR5 with broad-spectrum anti-human immunodeficiency virus type 1 activity. Antimicrob Agents Chemother. 2005;49(11):4721-32.
117. Fatkenheuer G, Pozniak AL, Johnson MA, Plettenberg A, Staszewski S, Hoepelman AI, et al. Efficacy of short-term monotherapy with maraviroc, a new CCR5 antagonist, in patients infected with HIV-1. Nat Med. 2005;11(11):1170-2.
118. MacArthur RD, Novak RM. Reviews of anti-infective agents: maraviroc: the first of a new class of antiretroviral agents. Clin Infect Dis. 2008;47(2):236-41.
119. Poveda E, Seclen E, Gonzalez Mdel M, Garcia F, Chueca N, Aguilera A, et al. Design and validation of new genotypic tools for easy and reliable estimation of HIV tropism before using CCR5 antagonists. J Antimicrob Chemother. 2009;63(5):1006-10.
120. Jaskolski M, Alexandratos JN, Bujacz G, Wlodawer A. Piecing together the structure of retroviral integrase, an important target in AIDS therapy. FEBS J. 2009;276(11):2926-46.
121. Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51(18):5843-55.
122. Sato M, Motomura T, Aramaki H, Matsuda T, Yamashita M, Ito Y, et al. Novel HIV-1 integrase inhibitors derived from quinolone antibiotics. J Med Chem. 2006;49(5):1506-8.
123. Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/ GS-9137). J Virol. 2008;82(2):764-74.
124. Chun TW, Stuyver L, Mizell SB, Ehler LA, Mican JA, Baseler M, et al. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc Natl Acad Sci U S A. 1997;94(24):13193-7.
125. Montaner JS, Lima VD, Harrigan PR, Lourenco L, Yip B, Nosyk B, et al. Expansion of HAART coverage is associated with sustained decreases in HIV/AIDS morbidity, mortality and HIV transmission: the “HIV Treatment as Prevention” experience in a Canadian setting. PLoS One. 2014;9(2):e87872.
126. Hsu MC, Schutt AD, Holly M, Slice LW, Sherman MI, Richman DD, et al. Inhibition of HIV replication in acute and chronic infections in vitro by a Tat antagonist. Science. 1991;254(5039):1799-802.
127. Cupelli LA, Hsu MC. The human immunodeficiency virus type 1 Tat antagonist, Ro 5-3335, predominantly inhibits transcription initiation from the viral promoter. J Virol. 1995;69(4):2640-3.
128. Hamy F, Felder ER, Heizmann G, Lazdins J, Aboul-ela F, Varani G, et al. An inhibitor of the Tat/TAR RNA interaction that effectively suppresses HIV-1 replication. Proc Natl Acad Sci U S A. 1997;94(8):3548-53.
129. Hwang S, Tamilarasu N, Kibler K, Cao H, Ali A, Ping YH, et al. Discovery of a small molecule Tat-trans-activation-responsive RNA antagonist that potently inhibits human immunodeficiency virus-1 replication. J Biol Chem. 2003;278(40):39092-103.
130. Hui B, Xia W, Li J, Wang L, Ai J, Geng M. Sulfated polymannuroguluronate, a novel anti-acquired immune deficiency syndrome drug candidate, blocks neuroinflammatory signalling by targeting the transactivator of transcription (Tat) protein. J Neurochem. 2006;97(2):334-44.
131. Lu CX, Li J, Sun YX, Qi X, Wang QJ, Xin XL, et al. Sulfated polymannuroguluronate, a novel anti-AIDS drug candidate, inhibits HIV-1 Tat-induced angiogenesis in Kaposi’s sarcoma cells. Biochem Pharmacol. 2007;74(9):1330-9.
132. Barbaro G, Scozzafava A, Mastrolorenzo A, Supuran CT. Highly active antiretroviral therapy: current state of the art, new agents and their pharmacological interactions useful for improving therapeutic outcome. Curr Pharm Des. 2005;11(14):1805-43.
133. Daelemans D, Pannecouque C. HIV-1 Rev function as target for antiretroviral drug development. Curr Opin HIV AIDS. 2006;1(5):388-97.
134. Flexner C. HIV drug development: the next 25 years. Nat Rev Drug Discov. 2007;6(12): 959-66.
135. Tedbury PR, Freed EO. HIV-1 gag: an emerging target for antiretroviral therapy. Curr Top Microbiol Immunol. 2015;389:171-201.
136. Fujioka T, Kashiwada Y, Kilkuskie RE, Cosentino LM, Ballas LM, Jiang JB, et al. Anti-AIDS agents, 11. Betulinic acid and platanic acid as anti-HIV principles from Syzigium claviflorum, and the anti-HIV activity of structurally related triterpenoids. J Nat Prod. 1994;57(2):243-7.
137. Li F, Goila-Gaur R, Salzwedel K, Kilgore NR, Reddick M, Matallana C, et al. PA-457: a potent HIV inhibitor that disrupts core condensation by targeting a late step in Gag processing. Proc Natl Acad Sci U S A. 2003;100(23):13555-60.
138. Smith PF, Ogundele A, Forrest A, Wilton J, Salzwedel K, Doto J, et al. Phase I and II study of the safety, virologic effect, and pharmacokinetics/ pharmacodynamics of single-dose 3-o-(3’,3’-dimethylsuccinyl)betulinic acid (bevirimat) against human immunodeficiency virus infection. Antimicrob Agents Chemother. 2007;51(10):3574-81.
139. Jacque JM, Stevenson M. The inner-nuclear-envelope protein emerin regulates HIV-1 infectivity. Nature. 2006;441(7093):641-5.
140. Shun MC, Daigle JE, Vandegraaff N, Engelman A. Wild-type levels of human immunodeficiency virus type 1 infectivity in the absence of cellular emerin protein. J Virol. 2007;81(1):166-72.
141. Mansharamani M, Graham DR, Monie D, Lee KK, Hildreth JE, Siliciano RF, et al. Barrier-to-autointegration factor BAF binds p55 Gag and matrix and is a host component of human immunodeficiency virus type 1 virions. J Virol. 2003;77(24):13084-92.
142. Van Maele B, Busschots K, Vandekerckhove L, Christ F, Debyser Z. Cellular co-factors of HIV-1 integration. Trends Biochem Sci. 2006;31(2):98-105.
143. Gallant JE, Koenig E, Andrade-Villanueva J, Chetchotisakd P, DeJesus E, Antunes F, et al. Cobicistat versus ritonavir as a pharmacoenhancer of atazanavir plus emtricitabine/ tenofovir disoproxil fumarate in treatment-naive HIV type 1-infected patients: week 48 results. J Infect Dis. 2013;208(1):32-9.
144. Cahn P, Andrade-Villanueva J, Arribas JR, Gatell JM, Lama JR, Norton M, et al. Dual therapy with lopinavir and ritonavir plus lamivudine versus triple therapy with lopinavir and ritonavir plus two nucleoside reverse transcriptase inhibitors in antiretroviral-therapy-naive adults with HIV-1 infection: 48 week results of the randomised, open label, non-inferiority GARDEL trial. Lancet Infect Dis. 2014;14(7):572-80.
145. Reynes J, Trinh R, Pulido F, Soto-Malave R, Gathe J, Qaqish R, et al. Lopinavir/ritonavir combined with raltegravir or tenofovir/ emtricitabine in antiretroviral-naive subjects: 96-week results of the PROGRESS study. AIDS Res Hum Retroviruses. 2013;29(2):256-65.
146. Raffi F, Babiker AG, Richert L, Molina JM, George EC, Antinori A, et al. Ritonavir-boosted darunavir combined with raltegravir or tenofovir-emtricitabine in antiretroviral-naive adults infected with HIV-1: 96 week results from the NEAT001/ANRS143 randomised non-inferiority trial. Lancet. 2014;384(9958):1942-51.
147. Greig SL, Deeks ED. Abacavir/dolutegravir/lamivudine single-tablet regimen: a review of its use in HIV-1 infection. Drugs. 2015;75(5):503-14.
148. Manzardo C, Gatell JM. Stribild(R) (elvitegravir/cobicistat/emtricitabine/tenofovir disoproxil fumarate): a new paradigm for HIV- 1 treatment. AIDS Rev. 2014;16(1):35-42.
149. Group ISS, Lundgren JD, Babiker AG, Gordin F, Emery S, Grund B, et al. Initiation of Antiretroviral Therapy in Early Asymptomatic HIV Infection. N Engl J Med. 2015;373(9):795-807.
150. Sturt AS, Dokubo EK, Sint TT. Antiretroviral therapy (ART) for treating HIV infection in ART-eligible pregnant women. Cochrane Database Syst Rev. 2010;(3):CD008440.
151. Grant RM, Lama JR, Anderson PL, McMahan V, Liu AY, Vargas L, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363(27):2587-99.
152. Montaner JS, Hogg R, Wood E, Kerr T, Tyndall M, Levy AR, et al. The case for expanding access to highly active antiretroviral therapy to curb the growth of the HIV epidemic. Lancet. 2006;368(9534):531-6.
153. Wood E, Kerr T, Marshall BD, Li K, Zhang R, Hogg RS, et al. Longitudinal community plasma HIV-1 RNA concentrations and incidence of HIV-1 among injecting drug users: prospective cohort study. BMJ. 2009;338:b1649.
154. Donnell D, Baeten JM, Kiarie J, Thomas KK, Stevens W, Cohen CR, et al. Heterosexual HIV-1 transmission after initiation of antiretroviral therapy: a prospective cohort analysis. Lancet. 2010;375(9731):2092-8.
155. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, et al. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N Engl J Med. 2009;361(23):2209-20.
156. Cohen T, Corbett EL. Test and treat in HIV: success could depend on rapid detection. Lancet. 2011;378(9787):204-6.
157. Granich RM, Gilks CF, Dye C, De Cock KM, Williams BG. Universal voluntary HIV testing with immediate antiretroviral therapy as a strategy for elimination of HIV transmission: a mathematical model. Lancet. 2009;373(9657):48-57.
158. Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278(5341):1295-300.
159. Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med. 1999;5(5):512-7.
160. Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. Proc Natl Acad Sci U S A. 1998;95(15):8869-73.
161. Karn J. The molecular biology of HIV latency: breaking and restoring the Tat-dependent transcriptional circuit. Curr Opin HIV AIDS. 2011;6(1):4-11.
162. Havlir DV, Marschner IC, Hirsch MS, Collier AC, Tebas P, Bassett RL, et al. Maintenance antiretroviral therapies in HIV infected patients with undetectable plasma HIV RNA after triple-drug therapy. AIDS Clinical Trials Group Study 343 Team. N Engl J Med. 1998;339(18):1261-8.
163. Blankson JN, Gallant JE, Quinn TC, Bartlett JG, Siliciano RF. Loss of HIV-1-specific immunity during treatment interruption in 2 chronically infected patients. JAMA. 2002;288(2):162-4.
164. Looney D, Ma A, Johns S. HIV therapy-the state of art. Curr Top Microbiol Immunol. 2015;389:1-29.
165. Pham QD, Wilson DP, Law MG, Kelleher AD, Zhang L. Global burden of transmitted HIV drug resistance and HIV-exposure categories: a systematic review and meta-analysis. AIDS. 2014;28(18):2751-62.
166. Médecins Sans Frontières. Untangling the web of antiretrovirals price reductions. 17th Ed. Geneva: MSF Access Campaign; 2014.
167. Waning B, Diedrichsen E, Moon S. A lifeline to treatment: the role of Indian generic manufacturers in supplying antiretroviral medicines to developing countries. J Int AIDS Soc. 2010;13:35.
168. Granich R, Gupta S, Hersh B, Williams B, Montaner J, Young B, et al. Trends in AIDS Deaths, New Infections and ART Coverage in the Top 30 Countries with the Highest AIDS Mortality Burden; 1990-2013. PLoS One. 2015;10(7):e0131353.
Received in August, 2015.
Accepted in November, 2015.
Carlos A Duarte. Grupo de Terapia de VIH, Departamento de Farmacéuticos, Centro de Ingeniería Genética y Biotecnología, CIGB. Ave. 31 e/158 y 190, Cubanacán, Playa, CP 11600, La Habana, Cuba.


{kind=link}
{kind=link}
{kind=link}
{kind=link}