










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La morbilidad pediátrica la determina una diversidad de enfermedades, entre las que no es común el síndrome neurocutáneo, sin embargo, las afecciones que este incluye afectan la calidad de vida de los afectados y la funcionalidad de su familia, lo que hace necesario su diagnóstico y tratamiento precoz.
El síndrome neurocutáneo, además denominado como hamartoblastosis, es un grupo de enfermedades con características clínicas y patológicas de diversa complejidad, que derivan de un trastorno disgenético que resulta en el desarrollo displásico de estructuras derivadas de las capas germinales embrionarias, principalmente del neuroectodermo.1
Entre las hamartoblastosis se reconoce el síndrome de Sturge-Weber (SSW)2, cuyo diagnóstico inusual y lo complejo de su tratamiento, motiva el objetivo de este artículo, dirigido a socializar la experiencia en la atención a un paciente pediátrico atendido en el Hospital Pediátrico Docente “General Pedro Agustín Pérez” y a presentar una revisión de la literatura para transmitir a la comunidad médica, de modo particular, a los estudiantes de medicina y médicos generales, información que los prepare para lograr un diagnóstico y un seguimiento adecuados de esta afección.
Presentación del caso
Paciente de un año de edad, procedencia área rural, nacido por parto vaginal a las 38 semanas, con buena adaptación neonatal, normopeso, sin antecedentes prenatales, ni posnatales, ni antecedentes familiares de interés, esquema de inmunización completo hasta el momento del ingreso. Al mes de edad fue operado de glaucoma congénito. Durante el primer año de vida presentó retardo del desarrollo psicomotor.
A los 7 meses de edad, los padres lo llevaron al cuerpo de guardia del Hospital Pediátrico Docente “General Pedro Agustín Pérez” de Guantánamo, por presentar movimientos involuntarios, que se consideraron como convulsiones tónico-clónicas, localizadas en hemicuerpo derecho, asociado a una pérdida de la conciencia y relajación de esfínteres, por lo que se decidió su ingreso para estudio y tratamiento.
Examen físico general
Piel: presentó mácula facial en vino de Oporto a nivel frontoorbitario derecho de aproximadamente 5 cm de diámetro, con bordes irregulares, sin presencia de vello cutáneo o elevación de la misma. También de forma similar, en las regiones pectoral y escapular del mismo lado, sugerente de hemangioma. (Figura 1)

Examen físico por aparato
Sistema Nervioso Central (SNC): paciente alerta, estableció contacto visual, intentó agarrar objetos, sin asimetrías faciales, tono muscular adecuado, postura normal, reflejos para la edad sin alteraciones. No se observaron movimientos anormales o signos meníngeos. Retardo en el neurodesarrollo, hemiparesia izquierda espástica, fondo de ojo (ojo derecho: disco definido, excavación 0,5.-0,6, vasos estrechos, retina no alteraciones; ojo izquierdo: normal).
Estudios complementarios realizados
Hemograma, enzimas hepáticas, pancreáticas, función renal y coagulograma, serología (VDRL y VIH), estudio de líquido cefalorraquídeo, ecocardiograma, radiografía de tórax: todos fueron normales.
Tomografía computarizada simple de cráneo (TMC). (Figura 2)

Fig. 2 Tomografía computarizada simple de cráneo: obsérvese imagen hiperdensa de 45 UH, no homogénea, localizada en la región frontal derecha que dibuja los surcos (calcificaciones), atrofia cortical frontal y corteza cerebral subyacente retraída, discrepancias en el tamaño de los ventrículos laterales.
Resonancia magnética nuclear (RMN). (Figura 3)
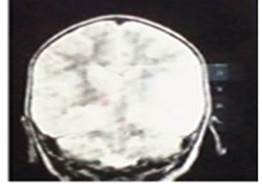
Fig. 3 Resonancia magnética nuclear: corte axial, obsérvese en hemisferio cerebral derecho atrofia cortical, espacios subacracnoideos dilatados y dilatación del sistema ventricular.
De acuerdo con las manifestaciones clínicas, el examen físico del paciente mediante el método clínico y los resultados de los exámenes complementarios se planteó el diagnóstico de síndrome neurocutáneo (SSW). Se aplicó tratamiento con diazepam a dosis de 0,25-0,50 mg/dosis 3 dosis y al persistir la crisis, fenitoína (bb 250 mg) a dosis de 18 mg/kg, luego, se prescribió a 6mg/kg/24 horas en 3 subdosis si convulsión. En las siguientes 48 horas el paciente evolucionó favorablemente. Luego se impuso tratamiento y egresó con valproato de sodio jarabe (250 mg - 5cc) a 25mg/kg/día.
Se orientó a la madre sobre la situación de salud de su hijo y como cooperar en su atención, afirmó entenderla y se comprometió a continuar con el seguimiento clínico periódico dado que ha evidenciado estabilidad y grandes avances en la salud de su hijo gracias al apoyo de un adecuado equipo multidisciplinario.
Discusión del caso
El paciente que se estudió presentaba manifestaciones cutáneas (angioma cutáneo) y neurológicas (hemiparesia y calcificaciones cerebrales), por lo que se planteó un síndrome neurocutáeno, de modo particular, un SSW, diagnóstico que se sustenta en los criterios que se revelan en la literatura científica.3,4
La primera clasificación de los síndromes neurocutáneos la realizó Jan Van der Hoeve, que acuñó el término facomatosis (phako = mancha) en 1932, en las que incluyó cuatro entidades: neurofibromatosis, esclerosis tuberosa y síndrome de Von Hippel Lindau, luego, se introdujo el término hamartoblastosis al incluir el SSW.4
Aproximación clínica al diagnóstico y manejo del síndrome de Sturge-Weber
El SSW lo describió en 1860 Rudolf Schirmer, quien asoció el angioma facial con el buftalmos. Luego en 1879, William Allen Sturge reconoció esta asociación a compromiso neurológico en esos mismos pacientes. En 1922, Frederick Parkes Weber demostró las alteraciones radiográficas que le caracterizan.5) También se conoce como angiomatosis meníngeo cutánea, angiomatosis encefalofacial, angiomatosis encefalotrigeminal, angiomatosis meníngea capilar, angiomatosis leptomeníngea, síndrome de Sturge Kalischer Weber, facomatosis de Sturge Weber, enfermedad de Dimitri y síndrome de Sturge Weber Dimitiri.
Epidemiología
Es un trastorno neurológico congénito, no hereditario aunque hay descrito casos familiares, poco común pero frecuente respecto a otros síndromes neurocutáneos. Se estima una incidencia de 5 x 100 mil nacidos vivos. Afecta a todos los grupos étnicos y ambos sexos.5
Etiología
Se atribuye a una mutación en el mosaico somático en GNAQ (c.548G > A, p.R183Q) en lesiones cerebrales y cutáneas. El GNAQ codifica Gαq, una sub-unidad alfa de proteínas G heterotriméricas.6,7
Fisiopatología
Los estudios histológicos evidencian un aumento en el número de vasos leptomeníngeos, atrofia y calcificación de la corteza. A medida que la atrofia cerebral se desarrolla, el número de vasos corticales disminuye, altera la perfusión cerebral y genera gliosis y pérdida neuronal.8,9 La atrofia cortical progresiva y la calcificación resultan de la isquemia cerebral. Las convulsiones prolongadas generan más daño isquémico y da lugar a un deterioro psicomotor.9,10) Se involucra en el mecanismo de la convulsiones un exceso de glutamato (GLU) liberado debido a la hipoxia crónica.10
Diagnóstico positivo
El diagnóstico se sospecha a partir del examen clínico para demostrar la presencia del angioma facial y la angiomatosis leptomeníngea, y se confirma mediante la TAC y la RMN.
Las manifestaciones clínicas se han definido con la triada diagnostica clásica: malformación capilar facial (mancha en vino de Oporto [MVO]), anomalías vasculares intracraneales (angiomas leptomeníngeos) y glaucoma.2
Las manifestaciones cutáneas se expresan por la presencia de angiomas faciales, una lesión macular secundaria a malformaciones capilares desordenadas, de paredes delgadas y dilatadas, ubicadas en la dermis y el tejido subcutáneo, presente al nacimiento, puede ser rosada en su inicio y con la edad oscurece hasta alcanzar un color rojo intenso (MVO)11,12, se localiza en la distribución dermatomal del nervio trigémino, usualmente V1 y V2. Se apunta que su distribución sigue los territorios de irrigación embrionaria más que los de inervación de un ramo del nervio trigémino.13
Las MVO aisladas son muy comunes, con mayor frecuencia localizadas en cara, cabeza y cuello8, pero pueden afectar a ambos lados del rostro y extenderse al tronco y las extremidades. Se estima que el 8 % de los pacientes con MVO congénita presentan SSW, cifra que aumenta a un 35 % si está presente en ambos lados de la cara.11) Su tamaño predice la gravedad neurológica de la enfermedad.14 Cuando se localiza en la frente o en el párpado superior, hay un riesgo de 10 a 35 % de presentar compromiso cerebral. Hay reportes de afectaciones como cutis marmorata telangiectásica generalizada.15
Las alteraciones vasculares afectan la boca y producen hiperplasia vascular de labios, mucosa oral, encías y área periodontal. Pueden ocurrir malformaciones vasculares viscerales en riñones, bazo, intestino, páncreas, pulmones y glándula tiroides.4
Las manifestaciones neurológicas incluyen cefalea, convulsiones, deterioro neurológico y cognitivo, alteraciones de la motilidad, retardo mental, trastornos del desarrollo y episodios de isquemia vascular con sus síntomas respectivos. Derivan de angioma leptomeníngeo presente hasta en el 98 % de los pacientes y es más común en la piamadre de los lóbulos occipital y parietal ipsilaterales a las MVO.16,17
Las convulsiones se presentan en el 75-90 % de los pacientes antes de los dos años de edad. Su severidad se relaciona con la extensión de la angiomatosis leptomeníngea y la progresión de la enfermedad. Al inicio suelen ser parciales, y en el tiempo evolucionan a crisis generalizadas Es menos común que se presenten como espasmos en flexión, mioclonías o crisis atónicas.18
La angiomatosis leptomeníngea condiciona isquemia cortical crónica, que genera áreas de calcificación, gliosis o atrofia cerebral.19) El 62 % de los niños tiene cefalea tipo migraña, de frecuencia variable, acompañada de auras, náuseas, vómitos, disartria, mareos y sensación de un pulso facial.19,20
El 33 % de los pacientes presenta hemiparesia o hemiplejía por oclusión venosa o isquemia de los tejidos locales, suele ser transitoria, pero la recurrencia conduce a hemiplejia persistente.19 Puede presentarse hidrocefalia y hemorragia intracraneal secundarias al deterioro del drenaje venoso cerebroespinal.4
Un 60 % de niños tiene retraso del desarrollo, problemas de aprendizaje y del comportamiento. En estos son más frecuente los trastornos del espectro autista y de oposición desafiante, delirios y alucinaciones, que se relacionan con la extensión de la lesión cerebral y con la gravedad y edad de inicio de las convulsiones.19,20,21
En el ojo, los angiomas producen glaucoma, hemangioma epiescleral o conjuntival, hemangioma coroidal, heterocromía o neovascularización del iris, hemianopsia homónima. El glaucoma se desarrolla en el 30-48 % de los pacientes, el 60 % es diagnosticado dentro de los primeros dos años de vida.4
A continuación se sintetizan los criterios para diagnóstico clínico del SSW:
Manifestaciones cutáneas: nevo flámeo o MVO, por lo general afectando la primera rama del nervio trigémino, presente desde el nacimiento, asociado con angiomas de las meninges y corteza cerebral del mismo lado.
Alteraciones neurológicas: calcificaciones cerebrales (consideradas patognomónicas del proceso), epilepsia, parálisis, paresias o parestesias en el hemicuerpo contra-lateral a la malformación vascular y retraso mental.
Alteraciones oculares: tumores vasculares de las coroides, conjuntiva y epiesclerales, glaucoma congénito o temprano, hemianopsia homónima, aumento del tamaño del ojo (buftalmus) y ceguera.
Alteraciones bucales del mismo lado que la MVO: hipertrofia del hueso maxilar, hiperplasia vascular y gingival que ocasionan maloclusión oral y asimetría facial.
Se señala como frecuentes las enfermedades infecciosas ópticas y nasofaríngeas, mayor el riesgo de apnea obstructiva del sueño y de complicaciones endocrinas (déficit de la hormona de crecimiento, hipopituitarismo e hipotiroidismo).4
El SSW, se clasifica como SSW completo e incompleto. De acuerdo a la escala de Roach se clasifica en los siguientes tipos:4
Tipo I: presencia de angioma facial y en leptomeninges, puede tener glaucoma (es la afección neuro-oculocutánea que define la forma completa, clásica de SSW).
Tipo II: presencia solo de angioma, sin daño del SNC afectado, puede tener glaucoma (presencia de angioma facial y glaucoma sin angioma leptomeníngeo, define la forma bisintomática de SSW).
Tipo III: presencia solo de angioma de leptomeninges, no glaucoma (presencia de angioma leptomeníngeo, sin involucrar la piel y el ojo, define la forma frusta de SSW).
Exámenes complementarios para el diagnóstico del síndrome de Sturge-Weber
La radiografía simple de cráneo evidencia las calcificaciones y la angiografía permite observar la ausencia de venas corticales superficiales.4) En la actualidad, la RMN cerebral con contraste de gadolinio es el examen de elección. La imagen típica es el angioma leptomeníngeo, que se traduce por captación de contraste en la duramadre en las secuencias T1. Otras imágenes típicas son la dilatación del plexo coroideo, calcificación y atrofia corticales, y dilatación del sistema venoso profundo. Las lesiones cerebrales son ipsolaterales al angioma facial.12,21) Si no está disponible la RMN, la TMC simple de cráneo identifica calcificaciones cerebrales y proporcionar alguna información anatómica.
La TMC por emisión de fotón único revela una disminución del flujo en la región cerebral con angiomatosis leptomeníngea21, y puede mostrar una disminución en la utilización de la glucosa en el hemisferio cerebral afectado. Esta tecnología ayuda a la identificación de los pacientes que pueden beneficiarse más de la intervención quirúrgica.4,17) Otros hallazgos que pueden ser encontrados en estos pacientes, son la atrofia de lóbulos o atrofia en otras localizaciones, hipertrofia de plexos coroideos, anomalías en el drenaje venoso, prominencias de las venas colaterales profundas, etc.4,17
No se encuentra un consenso sobre la edad óptima para el tamizaje en niños neurológicamente normales con angioma facial. En edades precoces puede no haber alteraciones de neuroimagen. Se sugiere que si el niño presenta un desarrollo normal, sin alteraciones en el examen neurológico ni convulsiones y tiene una imagen de RMN contrastada normal después del primer año de vida, probablemente no presente SSW con compromiso cerebral.22
A un niño con angioma facial en el territorio inervado por la primera rama trigeminal se le debe realizar RMN cerebral si surgen síntomas neurológicos, o a los 2 años de edad en los pacientes asintomáticos, si es normal en los lactantes sintomáticos, es necesario repetirla con 1-2 años de edad. Puede haber alteraciones de neuroimagen sin síntomas neurológicos, por lo que se debe realizar RMN en niños asintomáticos, si es normal a los 2 años de edad, no es necesario repetirla, excepto que aparezcan síntomas.22
Existen reportes de diagnósticos prenatales de SSW por medio de ecografía y RMN23) y la utilidad del doppler transcraneal en la valoración del flujo sanguíneo.24
En electroencefalograma (EEG), en el SSW, la actividad cerebral se atenúa y disminuye en el hemisferio afectado, esta asimetría se hace más evidente a medida que avanza la atrofia del hemisferio.25
El futuro del diagnóstico del SSW está en el desarrollo de biomarcadores, puede existir la posibilidad de diagnosticarlo mediante PCR digital de gotas ultra sensibles que detectan la mutación somática del GNAQ, no obstante, se necesitan más estudios.
Tratamiento
No hay una terapéutica específica para el SSW. Es necesario el manejo multidisciplinario del paciente para el monitoreo de la progresión de la enfermedad.
La MVO se trata con terapias láser, que debe comenzarse en la infancia, cuando es plana y rosada porque responde mejor, y puede disminuir la progresión y complicaciones que afectan la visión, las vías respiratorias y la deglución.15
El glaucoma se trata con timolol y el latanoprost, aunque puede ser necesario el tratamiento quirúrgico. Se ha utilizado el propranolol (oral) en el desprendimiento de retina por el hemangioma coroideo difuso. Se han reportado anecdóticamente tratamientos oftalmológicos antiangiogénicos.15
Se deben controlar las convulsiones para evitar el deterioro psicomotriz del paciente, generalmente se logra con 1 o 2 anticonvulsivantes más ácido acetilsalicílico. Los anticonvulsivantes más utilizados son oxcarbazepina, carbamazepina, levetiracetam y fenobarbital.25) Si aparecen espasmos infantiles se debe usar esteroides, topiramato, vigabatrina y la dieta cetogénica. En pacientes con convulsiones mioclónicas se debe utilizar valproato o levetiracetam. En aquellos con convulsiones refractarias, sobre todo si hay hemiparesia y déficit visual, se suele considerar la hemisferectomía, resección focal u otras intervenciones quirúrgicas11, en estos se ha evaluado el uso de cannabidiol.4,25
El ácido acetilsalicílico (3-5 mg/kg/ día) disminuye la frecuencia y la gravedad de las convulsiones.4 No está claro cuando comenzar el uso, ni se han evaluado otros fármacos antiplaquetarios o anticoagulantes.
La cefalea se puede prevenir con anticonvulsivantes, pues algunas se asocian a convulsiones. La lamotrigina es útil, pero se requiere un agente profiláctico.4,25
El seguimiento multidisciplinario y el apoyo psicosocial es importante en los pacientes y en sus familias. La adecuada orientación asegura la cooperación para cualquier tratamiento.
Pronóstico del paciente con síndrome de Sturge-Weber
El pronóstico depende de la extensión de la angiomatosis leptomeníngea y su efecto sobre la perfusión de la corteza cerebral, de la afectación ocular, el grado de afectación cerebral, la edad de inicio y el control de las convulsiones y si éstas logran ser controladas.12,21
Consideraciones finales
No siempre hay relación entre la gravedad de las manifestaciones cutáneas, neurológicas y oculares del SSW con las alteraciones cerebrales. Se connota la importancia del examen físico para establecer el diagnóstico oportuno para evitar futuras secuelas y complicaciones.
No se encuentra un consenso respecto a su manejo, si bien se reconoce las bondades del tratamiento del angioma facial con láser, los anticonvulsivantes, el tratamiento médico o quirúrgico para el glaucoma o cirugía funcional cerebral y el necesario manejo interdisciplinario de su atención profesional. Las investigaciones actuales se encaminan a la búsqueda de marcadores tempranos para predecir la progresión de la enfermedad.

