










 Servicios personalizados
Servicios personalizados Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkPresentación del caso
Paciente masculino de 31 años de edad producto de parto distócico a las 40,5 semanas de edad gestacional, talla 53 cm y peso 10,5 lb. Presentó un desarrollo psicomotor normal y dificultad en el lenguaje por tartamudez. Como antecedente familiar de interés, el abuelo paterno presentó tumor óseo de cráneo. A los 2 años le fue diagnosticada inmunodeficiencia humoral y celular por lo que recibió tratamiento; la evolución de la enfermedad fue satisfactoria. A los 14 años de edad se le detectó de forma incidental, en el curso de estudio odontológico, un tumor óseo en región frontoparietal izquierda; y posteriormente calcificaciones cerebrales en núcleos basales y sustancia paraventricular, así como otra lesión tumoral en fémur izquierdo. Ambos tumores fueron diagnosticados por estudio histopatológico como fibromixoma intraóseo. A los 19 años consulta nuevamente por presentar episodios de diarreas altas y cólicos abdominales. Se realiza biopsia yeyunal que mostró vellosidades de altura normal con cambios inflamatorios ligeros inespecíficos de la lámina propia. Ocho años después es valorado por la especialidad de neurocirugía por irritabilidad y episodios de cefalea. El electroencefalograma (EEG) reveló signos de irritación cortical focal. No hubo alteraciones en la resonancia magnética nuclear.
A los 30 años de edad presentó tres episodios de mareo intenso, rigidez del cuello y la boca, dificultad para hablar y tragar, desorientación y trastornos de percepción sin pérdida del conocimiento; además de: bajo rendimiento laboral, irritabilidad, parestesias, calambres, y debilidad muscular en miembros inferiores. Al examen físico presentó signos de tetania latente (Trousseau y Chvostek positivos), y fenotipo y somatometría normales. Los estudios analíticos mostraron hipocalcemia [Calcio (Ca) sérico: 0,98 mmol/L (VN: 2,05-2,75), Ca iónico: 0,51 mmol/L (VN:1-4)], hiperfosfatemia [fósforo (P): 1,93 mmol/L (VN: 0,68-1,8)] e hiperfosfatasemia [fosfatasa alcalina (FA): 314 UI (VN: 0-279)], los restantes parámetros analizados (glicemia, creatinina, ácido úrico, GGT, Na, K, Cl, VSG, hemograma y orina) siendo normales. Una segunda biopsia de yeyuno mostró atrofia parcial moderada de vellosidades intestinales con infiltrado linfoplasmocitario moderado en lámina propia y algunos eosinófilos, interpretado erróneamente como enfermedad celíaca y las crisis de tetania secundarias a esta entidad. Se suspendió el gluten de la dieta.
Se indicó tratamiento con calcio y magnesio parenteral con lo que se resolvió el cuadro agudo del paciente. La administración de calcio por vía oral tuvo poco efecto en la hipocalcemia.
Los valores de la parathormona (PTH) estuvieron marcadamente elevados [157,6 pg/mL (VN: 65-75)] y la hormona estimulante del tiroides (TSH) y tiroxina (T4) en rangos normales. La TAC de cráneo simple reveló múltiples calcificaciones en cerebro (cortical alta, lóbulos temporales y sustancia blanca pericallosa, más marcada en núcleos basales) y cerebelo. La densitometría ósea mostró signos de osteoporosis ligera de cadera derecha y osteopenia moderada de columna. En el ultrasonido de cuello se constató tiroides de tamaño y ecoestructura normal y paratiroides no demostrables. El ultrasonido abdominal fue normal y el EEG detectó signos de irritación cortical.
Seis meses después del cuadro clínico inicial se añadió calcitriol al tratamiento con calcio oral, y se observó una progresiva mejoría clínica y de los niveles de calcio y PTH.
Posteriormente, la biopsia de yeyuno fue reevaluada en el Instituto de Gastroenterologìa: la que arrojó el resultado siguiente: atrofia parcial ligera y parcial moderada de vellosidades duodenoyeyunales y atrofia parcial severa en uno de ocho fragmentos. Infiltrado linfoplasmocitario de ligero a moderado en la lámina propia. No aumento de linfocitos intraepiteliales. Se concluye “no enfermedad celíaca”.
El paciente es valorado de forma multidisciplinaria por las especialidades de Genética clínica, Gastroenterología y Endocrinología.
Discusión
Las causas más frecuentes de hipocalcemia severa, como la que presentó el paciente, son la deficiencia de vitamina D y de PTH, y la resistencia periférica a ellas.1)
La vitamina D se sintetiza en la piel a partir del precursor 7-dehidrocolesterol por acción de los rayos ultravioleta, o es absorbida de la dieta. Luego es transportada al hígado, donde se hidroxila a 25-hidroxi-vitamina D (25[OH]D). La conversión de 25[OH]D a 1,25-dihidroxi-vitamina D (1,25[OH]2D) o calcitriol, la forma activa de la vitamina D, ocurre en el túbulo renal, la cual es estimulada por la PTH. La principal acción biológica de la vitamina D es estimular el transporte activo de calcio y de fósforo a través del intestino. Por tanto, un déficit de esta se asocia a hipocalcemia e hipofosfatemia. Diferentes factores se asocian al déficit de vitamina D como son el déficit de exposición solar o de ingesta. Los cuadros de esteatorrea que se producen en algunos síndromes de malabsorción son la causa más común de déficit de vitamina D grave en caucásicos.1,2,3 Con relación al diagnóstico de enfermedad celíaca que se realizó inicialmente en este paciente, es de señalar que esta entidad, solo en estadios en que hay una marcada malabsorción con signos de desnutrición extrema, deshidratación hipotónica, y hemorragias debido a defecto en la síntesis de vitamina K, es que se presenta la tetania.4 Ambas biopsias de yeyuno no mostraron las alteraciones histopatológicas que caracterizan a esta enfermedad. Por otra parte, los cuadros de hipocalcemia crónica pueden llevar a síntomas digestivos como dolor abdominal y diarreas, lo que presentó el paciente en un momento evolutivo de la enfermedad.5 Otras entidades, como la enfermedad hepática severa, y la insuficiencia renal crónica interfieren en la producción de 25[OH]D y 1,25[OH]2D respectivamente. Los niveles normales de urea y creatinina en el paciente permitieron descartar la insuficiencia renal crónica Por otra parte condiciones genéticas como el raquitismo dependiente de vitamina D y raquitismo resistente a vitamina D conducen a hipocalcemia, aunque el comienzo de estas ocurre con más frecuencia en la infancia temprana. El caso que se presenta comenzó con el cuadro a los 14 años. Distintos medicamentos como los antineoplásicos y anticonvulsivantes, pueden llevar a una hipocalcemia lo cual fue descartado en este caso sobre la base de la historia clínica.1,2,3 Aunque no contamos con la cuantificación de vitamina D para el estudio de nuestro paciente, si pudimos determinar los niveles de fósforo los cuales estuvieron elevados, lo que nos hace pensar que la causa de la hipocalcemia no fue debida a un déficit de vitamina D.
El hipoparatiroidismo es un trastorno asociado a diferentes entidades y caracterizado por niveles bajos de PTH en presencia de hipocalcemia e hiperfosfatemia.6 En este paciente los niveles de PTH estaban elevados, lo cual descarta al hipoparatiroidismo como causa de hipocalcemia.
El seudohipoparatiroidismo (PHP) es un grupo de endocrinopatías raras que se caracterizan por resistencia a la PTH, hipocalcemia e hiperfosfatemia, hallazgos que fueron vistos en este caso.7,8 La PTH actúa uniéndose a un receptor de membrana acoplado a la subunidad α de la proteína G estimuladora (Gsα) encargada de activar la vía del monofosfato cíclico de adenosina (AMPc). Sus efectos consisten en incrementar la reabsorción de calcio a nivel del túbulo distal del riñón e inhibir la de fósforo en el proximal, estimular la síntesis de 1,25[OH]2D y liberar calcio del hueso.3,9
Se distinguen dos formas clínicas de PHP, el tipo I (PHP-I) y el tipo II (PHP-II), según la respuesta a la administración exógena de PTH medida en producción de AMPc urinario y de fosfato. En el PHP-I no hay respuesta de aumento de AMPc, mientras que en el PHP-II es normal. Sin embargo, la fosfaturia es deficiente en ambos PHP.7 El PHP-I puede ser clasificado a su vez en tres subtipos basados en: a) las características dismórficas de osteodisfrofia hereditaria de Albright (OHA) dadas por: (baja estatura, cara redonda, braquidactilia, obesidad, osificaciones subcutáneas y, en ocasiones, retraso mental) b) presencia o ausencia de resistencia a múltiples hormonas (tienen su mecanismo de señalización basado en la proteína G) y c) deficiencia en la actividad de Gsα medida según la respuesta de AMPc (in vitro) (tabla).10,11
Tabla Diagnóstico diferencial de los diferentes tipos de seudohipoparatiroidismo
| Ca | P | PTH | OHA | Resistencia plurihormonal | AMPc urinario tras PTH | Actividad Gsα | |
| PHP-Ia | ↓ | ↑ | ↑ | Sí | Sí | ↓ | ↓ |
| PHP-Ib | ↓ | ↑ | ↑ | No | No* | ↓ | N |
| PHP-Ic | ↓ | ↑ | ↑ | Sí | Sí | ↓ | N |
| PHP-II | ↓ | ↑ | ↑ | No | No | N | N |
P: fósforo; Ca: calcio; AMPc: monofosfato cíclico de adenosina; Gsα: subunidad α de la proteína G estimuladora;
N: normal; OHA: osteodistrofia hereditaria de Albright; PHP: seudohipoparatiroidismo; PTH: parathormona. ↓ disminuido; ↑ elevado
*La resistencia a la TSH ha sido reportada en algunos casos.8,12
Con la hipocalcemia detectada y la presencia de signos y síntomas relacionados con esta (tetania, irritabilidad, parestesias, calambres, debilidad muscular), hiperfosfatemia y elevación de la PTH, se realizó un diagnóstico presuntivo de PHP. En relación a las calcificaciones intracraneales se han descrito en hasta el 50 % de los pacientes con PHP, las cuales pueden estar asociadas o no a los rasgos físicos de OHA.13 Aunque en algunos pacientes con PHP-I se ha descrito enfermedad ósea (osteítis fibroquística) y un incremento en la prevalencia de exostosis óseas, la singularidad de este caso está en la asociación, no reportada en la literatura científica, del tumor tipo fibromixoma intraóseo con PHP.11,13
La ausencia del fenotipo de AHO y de resistencia plurihormonal permitió descartar los tipos PHP-Ia y Ic. Sin embargo, clínicamente no pudimos determinar si correspondía al PHP-Ib o II. La forma de aclarar el diagnóstico clínico es medir la respuesta del AMPc urinario a la administración de PTH sintética, la cual no fue posible realizar por no estar disponible en nuestro medio. Hasta el momento solo unos pocos casos de PHP-II han sido reportados y el defecto molecular aún no se conoce.7 Algunos autores han propuesto como hipótesis que el PHP-II pudiese ser un trastorno adquirido secundario al déficit de vitamina D, aunque esta teoría no ha sido corroborada.13 Para confirmar el diagnóstico se solicitó estudio molecular [Methylation specific_multiplex ligation-dependent probe amplification (MS-MLPA)] del gen GNAS (Guanine Nucleotide binding protein Alpha Stimulating), el cual mostró pérdida de metilación en los exones XLas, A/B y NESPas. La secuenciación de los 13 exones codificantes no presentó alteraciones compatibles con la clínica. Se confirmó el diagnóstico de PHP de tipo Ib.
El PHP es producido en la mayoría de los casos por alteraciones genéticas y/o epigenéticas en el locus GNAS localizado en el cromosoma 20q13, presentando defectos en la actividad de Gsα, necesaria para la señalización de hormonas tales como la PTH y la TSH.13,14,15 Estructuralmente el locus GNAS contiene 12 exones comunes (exones 2-13 de Gsα) que se unen a uno de los primeros cuatro exones alternativos. En función de qué primer exón alternativo se una a los exones 2-13 y se transcriba, se producirán cuatro tipos diferentes de proteínas o de transcritos. Estos incluyen la propia proteína Gsα, el transcrito no traducido A/B, y las proteínas XLαs y NESP55; asociados a los exones 1, A/B, XLαs y NESP55 respectivamente. Existe un transcrito antisentido no codificante llamado NESPas3,10,13 (Fig. ).
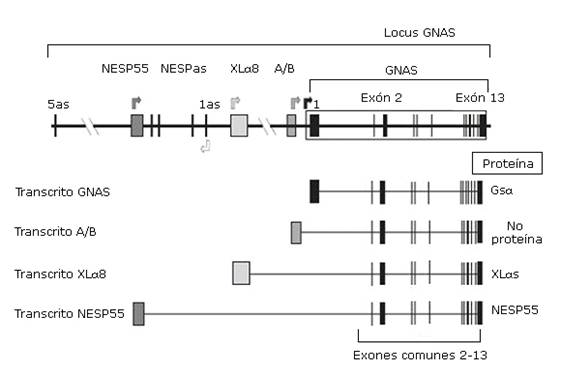
Fig Organización del locus GNAS. Las cajas y las líneas verticales representan los exones. Entre los exones (línea horizontal) están los intrones. Las flechas superiores indican el punto de comienzo de lectura de los cuatro transcritos principales. Los cuatro transcritos solo difieren en el primer exón 1 alternativo, mientras que los exones 2-13 son comunes. El exón 1, al unirse a los exones comunes 2-13, codifica para la propia proteína Gsα; el exón A/B, al unirse al resto de los exones comunes, genera un transcrito que no codifica para ninguna proteína; el exón XLαs, al unirse a los exones 2-13, codificará para la proteína XLαs; y el exón codificante de NESP55.10
Este gen presenta un complicado patrón de impronta, permitiendo la expresión de los distintos fenotipos en dependencia del sexo del progenitor que transmite el defecto molecular.9 En el establecimiento de la impronta (o inactivación de un alelo) la metilación del ADN (adición de grupos metilo en citosinas) desempeña un papel fundamental, constituyendo una de las modificaciones epigenéticas mejor estudiadas.10 En personas saludables los primeros exones alternativos del gen GNAS, referidos anteriormente, presentan el fenómeno de impronta y esta inactivación de uno de sus dos alelos varía según el transcrito. El transcrito NESP55 tiene impronta paterna (está metilado el alelo paterno), siendo expresado por el alelo materno, mientras que los exones XL XLαs, A/B y NESPas solo serán expresados por el alelo paterno.3,10) Los transcritos de Gsα se expresan bialélicamente, excepto en algunos tejidos como el túbulo renal proximal, tiroides, gónadas e hipófisis donde tiene impronta paterna. Se desconoce el mecanismo de impronta del exón 1 de GNAS.16
En cuanto al aspecto molecular el PHP-Ia y PHP-Ic presentan, por lo general, mutaciones en heterocigosis en cualquiera de los 13 exones del gen GNAS, y un patrón de herencia autosómica dominante.8,10 En relación a los pacientes con PHP-Ib, como es el caso que se presenta, muestran alteraciones en el patrón de metilación del gen GNAS.17 Puede presentarse de forma esporádica (casos aislados en una familia) o con un patrón de herencia autosómica dominante.18 La forma familiar se caracteriza por pérdida aislada de la metilación del exón A/B materno asociada a deleciones en el gen GNAS, o en el gen STX16, también heredadas de la madre en ambos casos.3,17) Debido al fenómeno de impronta, la resistencia hormonal solo aparece al heredar el defecto por vía materna, mientras que la transmisión del mismo por vía paterna no se asocia con anomalías endocrinológicas.13 En relación a los casos esporádicos, la perdida de metilación implica, además del exón A/B, a los otros exones alternativos.3,19,20 Las bases genéticas de esta variante se desconocen, excepto en algunos casos en quienes se ha reportado disomía uniparental paterna del cromosoma 20, lo cual significa que ambos cromosomas 20 han sido heredados del padre.21
La ausencia de historia familiar de hipocalcemia y resistencia a PTH, combinado con amplia pérdida de metilación del gen GNAS (exones XL XLαs, A/B y NESPas) y la no evidencia de deleciones, permitieron hacer el diagnóstico de una forma esporádica de PHP-Ib.
En relación con el asesoramiento genético, la descendencia de un individuo con PHP-Ib de tipo esporádico, como es el que se presenta, al haber pérdida de metilación combinada en varios exones, pero no detectarse deleciones (en caso de estar presentes pueden ser transmitidas con un 50 % de probabilidad, siendo las causantes de la pérdida de la metilación), el riesgo de transmisión de la enfermedad no puede definirse. Solo podemos decir que en el individuo estudiado la enfermedad está causada por la pérdida de metilación, pero el riesgo en las generaciones siguientes es incierto.10,11
Conclusiones
La alteración epigenética en el locus GNAS, en ausencia de expresiones fenotípicas, pero con hallazgos bioquímicos y clínicos permitieron realizar el diagnóstico de PHP-Ib de tipo esporádico y brindar un asesoramiento genético. Se presenta el primer reporte de esta enfermedad en Cuba con estudio molecular.

