










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El carcinoma de mama es la neoplasia maligna más frecuente en las mujeres. Ocurre cuando las células de la glándula mamaria crecen de forma descontrolada debido al fallo de los mecanismos regulatorios que controlan su proliferación. La incidencia de cáncer de mama (CM) se ha incrementado en un 50 % en las dos últimas décadas.1,2
Durante el 2012 se diagnosticaron, a nivel mundial, 1 677 000 casos de CM, lo que representa el 25 % de todos los cánceres. De modo que la incidencia mundial del cáncer de mama es de 38,9 % casos por cada 100 000 mujeres. Anualmente mueren en el mundo 400 000 mujeres por CM.3
Según el anuario estadístico, durante el 2018, el CM en Cuba se ubicó en el cuarto lugar con 1592 defunciones, precedido por tumores de tráquea, bronquios y pulmón; próstata e intestino. Tuvo una tasa de 28,2 por cada 100 000 habitantes. Mientras que, en el sexo femenino, ocupó el segundo nivel de prevalencia. El grupo etario más afectado por el carcinoma de mama se ubicó entre 60-79 años, lo que ratificó que, a mayor edad, mayor es el porciento de incidencia de esta afección.4
Uno de los problemas actuales de la oncología es la aparición de resistencias a un tratamiento, donde las células malignas se vuelven insensibles a los fármacos. Específicamente, con el uso de la genómica computacional, es posible la predicción de vías de señalización que se inhiben usando la terapia combinada de dos fármacos a partir de modelos in silico.5
Actualmente se avanza en el estudio de miles de genes que se expresan en un tejido tumoral y compararlo a su vez con otro sano para identificar los mecanismos moleculares involucrados. Se pretende, con una visión futurista, el establecimiento de un tratamiento personalizado que dependerá en cada caso del análisis y expresión de genes particulares en cada paciente. De ese modo, se podrá conocer la susceptibilidad a padecer algún tipo de cáncer, así como por qué dos tumores con características similares en diferentes personas pueden tener pronósticos desiguales.
El uso de las técnicas de biología molecular brinda una mejor comprensión acerca de los mecanismos implicados en el cáncer de mama. En ese sentido, el diseño in silico de nuevos fármacos ha permitido una mayor combinación de terapias no exploradas en el diseño experimental. Precisamente la bioinformática brinda las herramientas necesarias que hacen posible la selectividad y eficacia de una terapia, lo que reduce el tiempo y costo de la investigación presente.
Se conoce que las proteasas participan en procesos esenciales como: ovulación, fertilización, migración celular, inflamación, angiogénesis y apoptosis. Cambios en los patrones de expresión de las proteasas están involucrados en numerosos procesos patológicos que incluyen artritis, desórdenes neurodegenerativos, procesos inflamatorios y enfermedades cardiovasculares.6
La sobre expresión de la catepsina B, una proteasa lisosomal del tipo cisteíno, en los niveles transcripcionales y proteicos han sido observados en cáncer de esófago, gástrico, próstata, glioblastoma y cáncer de seno. Por ello, es considerada como un blanco potencial con propiedades terapéuticas debido a su amplia participación en la progresión del cáncer.6,7) Actualmente se trabaja en el diseño de inhibidores de las catepsinas y se ha observado una reducción en la tumurogénesis.
Sin embargo, con la llegada de la genómica computacional, se pueden explorar un mayor grupo de combinación de moléculas inhibidoras, diseñadas in silico, que constituyan blancos terapéuticos contra el CM. A continuación, se describen los aspectos estructurales y funcionales de la catepsina B, así como la selección de la mejor zona conservada que permita el diseño de inhibidores como candidatos potenciales en el tratamiento del carcinoma mamario.
MÉTODOS
Se consultó la opinión de expertos acerca de las terapias actuales empleadas contra el carcinoma de seno, así como sus principales ventajas y desventajas. Se procedió a la búsqueda de la proteasa catepsina B humana en las diferentes bases de datos online para la realización de su caracterización estructural y funcional. Las bases de datos consultadas fueron: InterPro (https://www.ebi.ac.uk/interpro/), Prosite (https://prosite.expasy.org/), Uniprot (https://www.uniprot.org/) y UniprotKB (https://www.uniprot.org/help/uniprotkb).
Seguidamente se procedió a la descarga de las secuencias proteicas de la catepsina B en formato FASTA encontradas en todos los organismos posibles a partir de la herramienta ENTREZ del NCBI (https://www.ncbi.nlm.nih.gov/), en la pestaña Protein. El fichero obtenido fue depurado usando un algoritmo de programación que permitía obtener todas las secuencias únicas que han sido anotadas, sin repetición, para un total de 2 940 secuencias. El fichero de texto en formato FASTA obtenido fue sometido a un alineamiento múltiple de secuencias con el programa on line Clustall Omega 1.2.4 en el sitio http://www.ebi.ac.uk/clustalw/index.html.
Los resultados otorgados por este programa necesitan ser analizados con un visualizador de secuencias. Para ello, se usó el visualizador Jalview 2.11.0, que le brinda por defecto, el programa Clustall Omega. Los resultados se mostraron a partir de la opción de Jalview que compara las secuencias proteicas según el por ciento de identidad de aminoácidos encontrado en cada una.
Posteriormente se eliminaron las secuencias no representativas, en unos casos, por ser muy cortas, y en otros, por no tener elementos aminoacídicos conservados, lo que resultó en una data de 2 485 secuencias de proteínas, que constituye una muestra representativa.
Seguidamente se procedió a la selección de las zonas de aminoácidos que se conservan. Se determinó la función de la zona conservada mediante las bases de datos disponibles.
RESULTADOS
Según los datos extraídos de la página del NCBI (National Center Biotechnology for Information), la catepsina B tiene una longitud de 339 aminoácidos, con un peso molecular de 37.363197 kD, su punto isoléctrico se encuentra a pH=5,46. Posee un coeficiente de extinción de 89400 M1cm-1 y un coeficiente de absorción de 2,39. La proteasa participa en múltiples procesos biológicos como la regulación de la actividad catalítica, proteólisis, regulación negativa de la muerte celular, procesos catabólicos del colágeno, actividad hidrolasa. Participa en la unión a proteoglicanos y complejos proteicos.
La catepsina B es una peptidasa cisteíno lisosomal de tipo papaína que se expresa en todos los tejidos y funciona primariamente como una exopeptidasa a través de su actividad carboxidipeptidil. Juntamente con otras catepsinas participa en el procesamiento de antígenos y activación de proenzimas. La actividad carboxipeptidil de esta proteasa se atribuye a la presencia de un lazo ocluyente en su sitio activo, lo cual favorece la unión del C-terminal de proteínas al sustrato. Se muestran las características aminoacídicas del sitio activo.(Fig. 1)

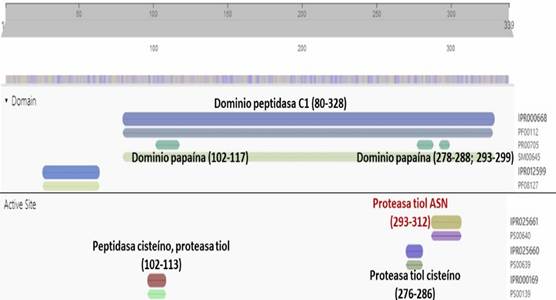
La base de datos UniprotKB adicionalmente agrega otros procesos en los cuales participa la catepsina B como la diferenciación celular, degranulación de neutrófilos, en la respuesta celular a hormonas tiroideas, vías de señalización de receptores tipo toll y la entrada viral en las células del hospedero. La proteasa preferencialmente cliva el enlace Arg-Arg-|-Xaa de sustratos pequeños. Por otro lado, la base de datos InterPro ofrece una caracterización estructural más detallada. (Fig. 2)
Se realizó el alineamiento múltiple de secuencias a partir de la herramienta Clustall Omega. Los fragmentos de los resultados obtenidos usando el visualizador Jalview. (Fig. 3)
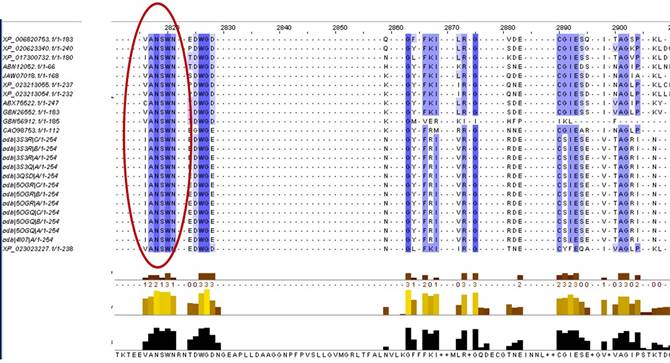
En color rojo se circula la zona mejor conservada de la catepsina B. Las barras de color negro muestran la probabilidad de encontrar cada aminoácido.
DISCUSIÓN
El alineamiento múltiple de secuencias de la catepsina B procedente de distintas especies. Los residuos de aminoácidos implicados en la catálisis de la familia de las peptidasa C1 incluyen una cisteína (C marcado en amarillo) en la posición 108 y una histidina (H marcado en amarillo) en la posición 278, que forman una diada catalítica. El otro residuo catalítico lo constituye una asparagina en la posición 298 (N marcado en amarillo) que orienta el anillo imidazol de la histidina catalítica. Otros residuos que juegan un papel importante en el mecanismo de acción de la proteasa es la glutamina (Q marcado en amarillo) que precede a la cisteína catalítica, se cree que ayuda en la formación de la cavidad del oxoanión. (Fig. 1)
A partir de los datos aportados por InterPro, se pudo determinar los tres dominios papaína de la catepsina B ubicados en las posiciones 102-117; 278-288 y 293-299, que incluye a su vez los tres residuos implicados en la catálisis. Adicionalmente se muestra la ubicación de los residuos catalíticos C 108, H 278 y N 298. La Cys y la His forman un par iónico el cual es estabilizado por la Asn mediante un puente de hidrógeno. Esta tríada tiene cierta similitud con el sitio activo de las serino proteasas (Ser, His, Asp). Sin embargo, en contraste con estas últimas, la cisteína nucleofílica está previamente ionizada a la unión del sustrato, razón por la que son vistas como enzimas activadas a priori.8
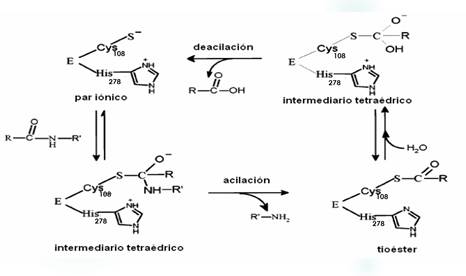
Durante la hidrólisis peptídica, el grupo tiol nucleofílico de la cisteína ataca el carbono carbonilo del enlace del esqueleto del sustrato a ser escindido y forma un intermediario tetraédrico que es estabilizado por la cavidad del oxianión. La secuencia de la reacción es mostrada, (Fig 4)
El intermediario tetraédrico se transforma en una acil -enzima (enzima-substrato tiol éster) con la simultánea libración de la porción C-terminal del sustrato (acilación). Este paso es seguido por la hidrólisis de la acil-enzima con agua, y se forma un segundo intermediario que finalmente se divide en la enzima libre,8 y la porción N-terminal del sustrato (deacilación).
La herramienta Clustal Omega ofrece el alineamiento múltiple realizado entre 2 485 secuencias de catepsina B. Con el objetivo de buscar las zonas que se conservan en la data, se analizaron todos los posibles resultados con el visualizador Jalview, y se escogió una zona de 5 residuos aminoacídicos que juegan un papel crucial en el mecanismo de acción de la peptidasa. El residuo conservado se muestra en color azul en la figura 3, circulado en rojo. La secuencia aminoacídica es ANSWN, que corresponde a los aminoácidos Alanina-Asparagina-Serina-Triptófano-Asparagina, ubicados en las posiciones 297, 298, 299, 300 y 301.
El análisis múltiple de secuencias incluye debajo de cada aminoácido, la presencia de barras en color negro que indican la probabilidad de encontrar dicho aminoácido en todas las secuencias analizadas de la proteasa. En este caso, la probabilidad de encontrar el aminoácido alanina en la posición 297 es de 70,42 %; la probabilidad de encontrar asparagina es de 95,68; serina, 82,57; triptófano, 94,33 y asparagina, 81,64. La asparagina ubicada en la posición 298 corresponde a uno de los residuos catalíticos que se conserva en todas las especies por la función crucial que tiene en el mecanismo de acción de la catepsina B.
La secuencia conservada (Alanina-Asparagina-Serina-Triptófano-Asparagina) incluye un aminoácido alifático, no polar (alanina). Su grupo metil, aunque es poco reactivo, participa en el reconocimiento de sustrato y especificidad. Incluye además dos asparaginas que son aminoácidos polares no cargados, cuyo grupo funcional se denomina carboxamida. Dicho grupo puede formar enlaces de hidrógeno con el esqueleto del péptido, los residuos de asparagina suelen encontrarse al principio y al final de la estructura de hélice alfa, y en vueltas o asas en láminas beta. La serina comparte propiedades estructurales similares a la asparagina pues resulta un aminoácido no cargado y polar por contener un grupo hidroxilo en la posición tres.
Constituye parte del sitio activo de muchas enzimas lo que determina su variabilidad funcional. su cadena lateral puede sufrir O-glicosilación, en la que puede haber una relación funcional con la diabetes. Es uno de los tres residuos de aminoácidos que son comúnmente fosforilados por las quinasas en la señalización celular en organismos eucariotas. El otro aminoácido contenedor en la secuencia conservada es el triptófano clasificacido como hidrofóbico aromático. Es esencial para promover la liberación del neurotransmisor serotonina, involucrado en la regulación del sueño y el placer.9
En este caso se trata de una secuencia ortóloga que se encuentra en diversas especies y se ha mantenido invariable lo que sugiere un patrón de conservación estructural que comparte funciones imprescindibles para el funcionamiento correcto de la catepsina B. Una de sus funciones comprende la participación en el sitio activo por parte de la asparagina 298. Por otro lado, presenta alta conservación en las especies a partir del por ciento de probabiblidad encontrado para cada aminoácido.
Este trabajo constituye la primera referencia en el trabajo inicial de búsqueda de inhibidores de catepsinas B a partir de secuencias conservadas evolutivamente en la proteína. Sin embargo, al presente se trabaja en el desarrollo de inhibidores de catepsinas B y L que se dividen en cinco grupos según su estructura: derivados de nitrilos, cetona, vinil sulfona, acilhidracina y estructuras químicas misceláneas. En ese sentido, se han obtenido buenos resultados en ensayos de actividad anti cáncer in vitro en el caso de las estructuras misceláneas, así como derivados de vinil sulfonato y cetonas en cáncer de mama metastásico.
Se observa que los derivados de nitrilos son los candidatos más atractivos por su potente inhibición, selectividad y reversibilidad, además de su baja reactividad hacia otros nucleófilos celulares.10
Se han reportado ciertos derivados de las calconas como inhibidores de la familia cisteíno proteasas. Por ello se ha profundizado en el efecto de algunas semicarbazonas, tiosemicarbazonas, pirazoles y pirazolinas como inhibidores de la actividad proteolítica endógena. Las calconas constituyen un grupo importante de productos naturales que son responsables de exhibir una amplia variedad de efectos farmacológicos incluyendo la actividad antitumoral, antimalaria, antiviral, antiinflamatoria y como agente anticancerígeno. Desde el punto de vista estructural, las calconas están compuestas en su núcleo central por una cetona aromática y una enona. Algunos de sus derivados conocidos como acil hidracidas y triazoles han sido reportados como inhibidores no peptídicos efectivos de las catepsinas B y H. Los inhibidores más potentes fueron los compuestos nitro sustituidos para la catepsina B y los compuestos cloro sustituidos para la catepsina H.11
Otro ensayo exhibe el diseño y síntesis de derivados de la nitrosolina como inhibidores de catepsinas B. Se omitió la parte piridina o la modificación de las posiciones 2,7 y 8 de la nitrosolina. Los compuestos fueron evaluados por su habilidad de inhibir la actividad endopeptidasa y exopeptidasa. Se determinó su habilidad para reducir la degradación intra y extracelular del colágeno lV en modelos in vitro y basados en células.12
Los inhibidores actuales de catepsinas carecen de actividad farmacológica y especificidad adecuada. Por ello, se trabaja en la obtención de un inhibidor de anticuerpo humanizado contra la catepsina B a través de la fusión del pro-péptido de la pro-catepsina B en la región tres determinante de la complementariedad de la cadena pesada de Herceptina. Este resultado, pro-pétido-anticuerpo tuvo una alta especificidad para inhibir la actividad proteolítica de catepsina B en niveles nanomolares.13
La inhibición de catepsina B puede dirigir la célula fuera de la muerte apoptótica hacia la necrosis, estimular una respuesta inmune y promover la inmunidad antitumoral. Se reporta la síntesis y caracterización de propiedades biológicas y fotoquímicas de inhibidore de catepsina B que son altamente selectivos y se activan rápidamente ante la exposición a la luz visible. El trabajo confirma que la inactivación de la catepsina B unido a la producción de oxígeno constituye un tratamiento promisorio para destruir las células tumorales.14 Mediante estudios de acoplamiento molecular (docking molecular) se modificaron nueve agente intercalantes en el ADN, derivados de aminoacridinas, debido a su acción antiproliferativa. Los compuestos poseen actividad inhibitoria contra HER2 y fueron seleccionados como candidatos potenciales antineoplásicos contra el CM.15
De igual modo, otros compuestos como miricetina, quercetina, apigenina, luteonina y baicaleína mostraron actividad anticancerígena contra la línea germinal MCF-7 para el cáncer de mama positivo al receptor a estrógenos. Ello demuestra las potencialidades actuales de los análisis in silico y herramientas bioinformáticas en el diseño de fármacos.16
Por esa razón se hace necesario la búsqueda de nuevos candidatos potenciales que disminuyan los efectos adversos de las terapias existentes contra el carcinoma mamario. En esa dirección, los inhibidores de catepsinas resultan una solución atractiva dentro del diseño in silico de fármacos. El uso de las técnicas de bioinformática reduce los costos y tiempo de la investigación, así como su contribución significativa a la bioseguridad de los inhibidores. En este trabajo, la selección de la zona conservada en la catepsina B, constituye un blanco terapéutico prominente en el desarrollo de medicamentos contra el cáncer de seno.

