










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El término ictiosis deriva de la palabra griega ichtys que significa pez, fue introducido por Williams en 1808 y define un grupo de trastornos generalizados de la cornificación, que se caracterizan por presentar hiperqueratosis y/o descamación. Su patogenia radica en la mutación de las proteínas esenciales para la formación de la queratina y la síntesis de lípidos.1,2
Entre las formas congénitas, la ictiosis arlequín es la forma más grave, pues causa gran mortalidad, los afectados rara vez sobreviven más allá de una semana.3 En 1750, Hart describió el primer caso, en el cual las escamas características en forma de rombo recordaban el traje del payaso arlequín, personaje clásico de la comedia italiana del siglo XV l.4
La clasificación más actual de la ictiosis distingue dos grandes formas: las no sindrómicas, que afectan exclusivamente la piel y las sindrómicas que se manifiestan en la piel y otros órganos. Las ictiosis congénitas autosómicas recesivas (ICAR) se encuentran dentro del grupo de las no sindrómicas, y en estas se incluyen un amplio espectro de fenotipos: la ictiosis laminar, el eritrodermia ictiosiforme, el bebe colodión e ictiosis arlequín.5 Las ICAR se observan en 1:138 000 - 1:300 000 nacidos.
Existen tres cuadros clínicos que se manifiestan en el recién nacido y se ha propuesto el término ictiosis congénita autosómica recesiva (conocidos como ARCI por sus siglas en inglés) para referirse a ellos: ictiosis arlequín, ictiosis lamelar y eritrodermia ictiosiforme congénita no ampollosa.6 Se reconoce ahora que estos fenotipos son parte de una misma entidad, y que las descripciones fenotípicas son útiles para determinar el pronóstico y el manejo de los individuos afectados.7
Ante el nacimiento de un neonato con características de arlequín las autoras se motivaron a realizar esta presentación de caso con el objetivo de profundizar en el conocimiento de estas genodermatosis en el recién nacido.
PRESENTACIÓN DEL CASO
Recién nacido masculino, de raza blanca, hijo de madre de 28 años de edad, con antecedentes patológicos familiares de cardiopatía congénita (hermana materna), con antecedentes de salud, con historia obstétrica de G4 A2 (provocados) P 1 (a término), serología no reactiva, grupo y factor a positivo, clasificada como fisiológica en la gestación actual. Con un tiempo gestacional de 34 semanas por fecha de última menstruación tras 1 h de trabajo de parto, con rotura prematura de membranas de 2 días, líquido amniótico claro se realiza cesárea por cesárea anterior en trabajo de parto extrayéndose recién nacido en presentación cefálica con un peso de 2 490 gramos, puntaje de Apgar de 6/8, cordón umbilical con una circular laxa al cuello, placenta normal y completa.
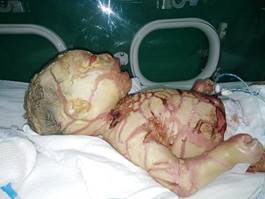
Se observa al examen físico piel gruesa, con grietas (Fig. 1), ectropión y eclabium, nariz plana con distensión de alas nasales (Fig. 2), dedos atróficos con amputación de falanges distales y deformidad en flexión de ambas manos y pies (Fig. 3). Es ingresado en la unidad de cuidados intensivos neonatales del Hospital Ginecobstétrico Provincial “José Ramón López Tabrane” con diagnóstico de ictiosis congénita. El paciente fue mantenido con hidratación endovenosa, cobertura antibiótica, oxigenoterapia y un ambiente estéril para evitar infecciones.
Los exámenes de laboratorio revelaron los siguientes hallazgos:
Hb 15,2 g/l.
Hto. 048.
Glicemia 6,0 mmol/L.
Proteina C Reactiva cualitativa positiva,
Recuento plaquetario 170x10-3/L.
Leucograma 4,1x109/L (seg 042, linf 048, Mon 009, Eo 000),
Gasometría arterial: ph 7,31; Pco2 39,1; PO2 125,9; EB -6,4; CHCO3 19,3.
A los 11 días de vida (Fig. 3) se produce un empeoramiento de su estado general con episodio de bradicardia y desaturación sin respuesta a las maniobras de reanimación y fallece.
Se realizó necropsia cuyo resultado informó: piel seca y acartonada con pérdida de epitelio, se observaron pulmones con un color amarillento y pálido, el corazón sin alteraciones, glándulas suprarrenales congestivas, hígado amarillo. Se observó esofagitis y gastritis. Riñón derecho con área de hemorragia en el polo superior; riñón izquierdo con dilatación calicial. Hallazgo necrópsico primario: Malformación congénita (ictiosis) y sepsis generalizada.
DISCUSIÓN
La primera descripción de IA fue hecha por el Reverendo Oliver Hart, de Charleston, Carolina del Sur en 1750.10 Este trastorno debe su nombre al aspecto de la piel que presentan los recién nacidos, caracterizada por escamas en forma de rombo, que recuerdan el traje del payaso arlequín, personaje clásico de la comedia en Italia en el siglo XVl.8
Aproximadamente 200 casos de ictiosis arlequín han sido publicados. Según la literatura científica para el 2007, se habían reportado 101 casos en todo el mundo en la literatura médica. La incidencia está calculada en 1 por 300.000 nacimientos.9 Se trata de una entidad de herencia autosómica recesiva, causada por la mutación del gen ABCA12 (adenosine triphosphatebinding cassette A12), que resulta en el transporte defectuoso de lípidos, alteración que impacta de manera significativa el desarrollo normal de la piel.10
Los bebés suelen nacer prematuros y están “encerrados” en un estrato córneo engrosado que a menudo se describe como armadura. Después del nacimiento se producen fisuras rojas en estas placas duras e inflexibles que se extienden hasta la dermis resultando en una piel parecida a un comodín. Presentan también microcefalia, ectropión y eclabio. El conducto auditivo externo y las fosas nasales aparecen rudimentarios.11 Además, tienen insuficiencia respiratoria como resultado de la expansión torácica restringida y deformidades esqueléticas. Se presentan problemas de alimentación, deshidratación e insuficiencia renal. Además, la inestabilidad térmica y las infecciones son comunes.11,12 En el caso presentado se observaron todas las características descritas.
El tratamiento temprano de la ictiosis arlequín está orientado al mantenimiento de la temperatura del recién nacido colocándolo en una incubadora con ambiente humidificado, cuidado de la piel y los ojos, soporte nutricional, control del dolor, fisioterapia y control de las infecciones.13 La mortalidad es muy alta y la mayoría de los neonatos afectados mueren en los primeros días debido a sepsis, fallo respiratorio, infecciones, pobre nutrición y desequilibrios hidroelectroliticos.14 Para el cuidado tópico de la piel se recomiendan baños diarios con jabones antisépticos y el uso de pomadas emolientes cada 4 a 6 h para mantener la piel suave e hidratada; se puede usar vaselina estéril o aceites inertes. La lubricación de la córnea en los casos con ectropión previene la sequedad de la córnea.15 En el neonato que se presentó se utilizó vaselina estéril aplicada cada 4 h.
Es importante realizar desde un inicio el diagnóstico adecuado para poder guiar a los padres en el conocimiento de la enfermedad. La atención multidisciplinaria y la implementación de un plan de cuidados, es esencial para el éxito del tratamiento. El tratamiento va encaminado a mejorar las características de la piel y mucosas y prevenir las infecciones. El diagnóstico prenatal y el consejo genético constituyen elementos fundamentales en el manejo de esta rara entidad.
Aunque en la práctica clínica es una entidad poco común, el personal de salud debe conocer las medidas iniciales de soporte para prevenir complicaciones y poder ofrecer un manejo multidisciplinario a estos pacientes.

