










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
La intubación endotraqueal se ha convertido en una práctica corriente dentro de la anestesiología moderna. Sin embargo, en ocasiones sorprende la dificultad o la imposibilidad de efectuarla aun en manos experimentadas. Por esta razón se hace necesaria la predicción de la dificultad o facilidad que ofrezcan las características anatómicas de cada paciente.1-4. En el niño, debido a la falta de cooperación al examen físico y de pruebas específicas que puedan predecir una vía respiratoria anatómicamente difícil, la evaluación de esta difiere de la del adulto. El abordaje de la vía respiratoria en los niños con anomalías craneofaciales es un reto para los anestesiólogos. Por tanto, se debe tener conocimiento sobre estas anomalías, como son el labio y paladar hendido con y sin Pierre-Robin,5 disostosis mandibulofacial, microsomia hemifacial, disostosis craneofacial, síndrome de Down, de Klippel-Feil, de Beckwith-Wiedemann, entre otras. El síndrome de Beckwith-Wiedemann (SBW) es una alteración con crecimiento exagerado que fue descrita en 1963 en Los Ángeles, California por J. Bruce Beckwith, quien informó tres casos posmortem y año después por Wiedemann en Kiel Alemania que reportó tres casos de hermanos.6,7 Ambos investigadores describieron características similares en sus pacientes que no correspondían a ninguno de los síndromes craneofaciales hasta entonces descritos. También se le conoce como síndrome de Onfalocele-Macroglosia-Gigantismo o EMG. Este síndrome es eventual, pero puede tener un carácter hereditario autosómico dominante con características clínicas muy variables. Tiene una incidencia de 1:15 000 nacimientos, aunque otros autores plantean que puede llegar a 1 de cada 13 700, cifras que son aproximadas ya que las formas leves no son diagnosticadas. Se considera que este síndrome se debe a una alteración genética, ya sea por una mutación nueva o por transmisión familiar en forma autosómica dominante.8,9 La incidencia en uno y otro sexo es similar y la frecuencia de presentación igual en individuos de la raza blanca o negra.10
Las características diagnósticas más comunes son macrosomia unilateral, macroglosia y onfalocele. Otras alteraciones que acompañan a este síndrome son hipoglucemia neonatal o infantil, alteraciones metabólicas, visceromegalias, exoftalmos, tumores embrionarios, retraso psicomotor, microcefalia, cardiopatías congénitas, hipoventilación alveolar, hipotiroidismo, policitemia y malformaciones óseas, entre otras. Por lo general no se encuentran cambios en el cariotipo y solo en 1 % hay anomalías en el cromosoma 11p15.5, 11p15.5, 11p15.5, 5q35.11 La existencia de las anomalías descritas y el trastorno metabólico son característicos de la enfermedad, por lo que la presencia de estos elementos permite hacer el diagnóstico positivo de esta. El diagnóstico diferencial debe establecerse con el síndrome de Simpson-GolabiBehmel, síndrome de Perlman, síndrome de Costello y con la hemihiperplasia aislada que puede asociarse con el síndrome de Proteus y de Klippel-Trenauny-Weber.
Estos pacientes pueden requerir intervención quirúrgica desde su nacimiento o en cualquier etapa de su vida pero la mayoría de los casos reportados han sido intervenidos antes de los tres años de edad. Las correcciones más frecuentes han sido el onfalocele, hernias inguinales y macroglosia. Otra intervención quirúrgica de menor frecuencia son las correcciones del labio y paladar hendido.12,13 Más de 50 % de estos pacientes nacen prematuros, lo cual incrementa su riesgo anestésico debido a las enfermedades propias de la prematurez como membrana hialina.14. Se ha descrito un lactante con este síndrome al que se le realizó resección parcial de lengua para facilitar su alimentación al seno materno.15
PRESENTACIÓN DEL CASO
Se trata de un paciente masculino mestizo de 4 años de edad, con peso 22 Kg, signos vitales dentro de parámetros normales para su edad, y antecedentes perinatales de polihidramnios, quiste renal derecho (hallazgo radiológico prenatal), macrosomia fetal y de hipoglucemia neonatal, sin antecedentes familiares del síndrome de Beckwith-Wiedemann (SBW), que es traído a la consulta preoperatoria con diagnóstico de tumor de Wilms para recibir tratamiento quirúrgico.
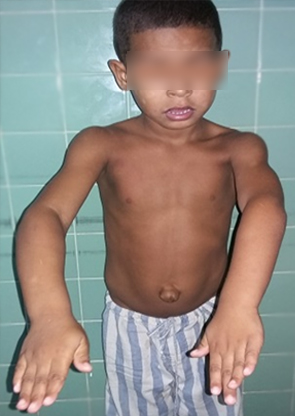
Los datos más relevantes del examen físico fueron: pliegues retroauriculares, macroglosia con asimetría, test de Mallampati Grado II, hernia umbilical, hipertrofia de hemicuerpo derecho. El laboratorio mostró Hb 127 g/L, glucemia 5,2 mmol/L, eritrosedimentación 25 mm/h, conteo de plaquetas 360×109/L, tiempo de coagulación 8' y tiempo de sangramiento 1½’, LDH 1307 U/L.
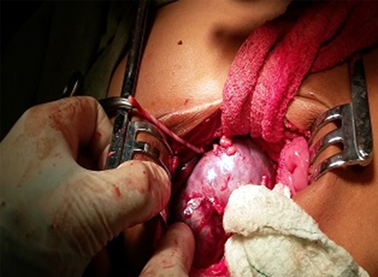
Se contó además con la valoración de un equipo multidisciplinario integrado por otorrinolaringólogos, cardiólogos, cirujanos, endocrinólogos e imagenólogos. En los estudios imagenológicos lo más significativo fue el ultrasonido renal: riñones aumentados de volumen para su edad, proceso tumoral ecogénico que ocupa el área de la suprarrenal derecha de más menos 72×63 mm de contorno polilobular y la presencia de calcificaciones en contacto con el polo superior del riñón, vena cava inferior desplazada hacia la línea media. La tomografía axial computarizada confirmó el proceso tumoral antes descrito.
Para el manejo de la vía respiratoria se incluyó mascarilla facial, cánulas de Guedel, mascarillas laríngeas 2.5 Clásica y Supreme, tubos endotraqueales 5,0 y 5,5 con y sin manguito y, laringoscopio de fibra óptica con espátulas curvas y rectas No. 2. Se premedicó con atropina 0,02 mg/kg, midazolam 0,07 mg/kg, metilprednisolona 4 mg/kg por vía intravenosa.
Una vez en el quirófano se canalizó otra vía periférica en miembro superior izquierdo calibre 22, se monitorizó tensión arterial no invasiva, electrocardiograma, oximetría digital, capnografía y temperatura. Se inició la inducción con ketamina 2 mg/kg, succinilcolina 1 mg/kg; acto seguido un anestesiólogo calificado realizó la laringoscopia directa para determinar la dificultad de la vía respiratoria, la cual mostró un grado II según la clasificación de Cormack˗Lehane, se coloca un tubo endotraqueal No. 5 con manguito por vía oral y la anestesia se mantuvo con Ο2/isoflurano, además de fentanilo y vecuronio. Se colocó sonda vesical, sonda nasogástrica, y se le realizó abordaje venoso subclavio derecho para la administración de soluciones coloides y cristaloides.
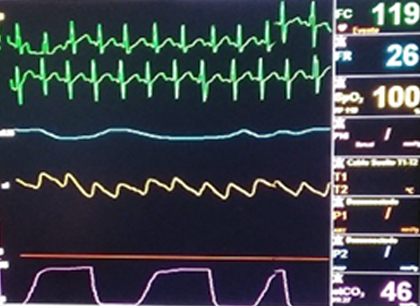
La intervención quirúrgica transcurrió sin accidentes y no hubo incidentes transanestésicos (Fig 1). Se realizaron gasometrías seriadas constatándose normoglucemia y no fue necesaria la administración de glóbulos (Fig 2). Fue trasladado a la unidad de cuidados intensivos pediátricos con ventilación espontánea (Fig. 3).
DISCUSIÓN
La literatura relacionada con este tipo de pacientes es muy escasa, por lo que el tratamiento quirúrgico es considerado un desafío para el anestesiólogo. La prematurez favorece una mortalidad hasta de 20 %, particularmente relacionada con hipoglucemia neonatal, cardiopatía y la macroglosia.15-17 Esta macroglosia y la macrosomia habitualmente se presentan desde el nacimiento, aunque se ha visto que ambas pueden desarrollarse en el período posnatal; a pesar de lo descrito, en este caso, la intubación no fue difícil y se pudo colocar el tubo orotraqueal con una clasificación de Cormack˗ Lehane Grado II, esto fue posible debido a que la lengua no era enorme. También se describen alteraciones usuales en estos individuos como el onfalocele, hernia umbilical, diastásis de rectos. El aumento volumétrico del hemicuerpo o hemihiperplasia se presenta desde el nacimiento y se hace más evidente durante su crecimiento. Este paciente no presentó anomalías anatómicas cardíacas por lo que se coincide con lo descrito por la literatura que asevera su infrecuencia. Viljoen y otros,18 en su estudio con 13 niños con este síndrome mostraron que 12 tuvieron anomalías cardiovasculares; 7 con cardiopatías congénitas (defectos septales interauriculares y ventriculares, persistencia del conducto arterioso, tetralogía de Fallot, hipoplasia ventricular izquierda), y 5 con cardiomegalia idiopática. También se ha descrito la malformación Chiare.19
Este paciente es operado de un tumor de Wilms, y se coincide con lo reportado por autores como Choyke y Borer20,21 quienes describieron en sus estudios la posibilidad de encontrar alteraciones renales como displasia medular, sistema colector duplicado, nefrocalcinosis, quistes, medula renal en esponja, divertículos, nefromegalia y tumor de Wilms.
A pesar de que la hipoglucemia ha sido mencionada como uno de los factores determinantes en la evolución de estos pacientes, en este caso no existió efecto adverso relacionado con este parámetro, el cual se monitoreó con la realización de glicemias, así como el aporte de dextrosa durante todo el proceder. Se han descrito otras alteraciones metabólicas menos frecuentes como el hipotiroidismo, hiperlipidemia, hipercolesterolemia, policitemia e hipercalciuria pero ninguna se presentó en el perioperatorio.22
La evaluación pediátrica preanestésica en imprescindible en estos pacientes. Para los anestesiólogos, los pacientes con síndrome de Beckwith-Wiedemann deben considerarse de alto riesgo debido a que son portadores de una vía respiratoria anatómicamente difícil, por tanto, debe realizarse una evaluación detallada de esta, valorar comorbilidades, tipo de cirugía planeada y estado clínico perioperatorio.

