










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.9 no.6 Cienfuegos nov.-dic. 2011
PRESENTACIÓN DE CASO
Síndrome frágil X, diabetes mellitus tipo 1 y síndrome de malabsorción intestinal
Fragile X Syndrome, Diabetes Mellitus Type 1 and Malabsorption Syndrome
Rafael Pila Pérez , Rafael Pila Peláez , Pedro Rosales Torres , Víctor Holguín Prieto
Hospital Universitario Manuel Ascunce Domenech, Camagüey, Camagüey, Cuba
RESUMEN
Se presenta el caso de un paciente de 28 años de edad con el diagnóstico de síndrome frágil X, diabetes mellitus tipo 1 y síndrome de malabsorción intestinal por giardias, entidad poco común y cuyas asociaciones constituyen el primer caso reportado en el país. Se reseñan algunas características genéticas de este síndrome y al mismo tiempo se exponen las principales características clínicas y diagnósticas, además se señala la conducta terapéutica empleada. Se concluye que el síndrome frágil X es uno de los trastornos hereditarios más comunes y que resulta desconocido por la población en general y los profesionales de la salud, por lo que se hace necesario su conocimiento para indicar el asesoramiento genético a la familia de pacientes con retraso mental. Ha motivado la realización de este trabajo el haber diagnosticado un paciente con SFX asociado a diabetes mellitus tipo 1 y síndrome de malabsorción secundario a giardiasis lo cual no ha sido reportado aún en Cuba ni en la literatura mundial.
Palabras clave: síndrome de cromosoma x frágil, diabetes mellitus tipo I, síndrome de malabsorción, retraso mental.
ABSTRACT
The case of a 28 years old patient with a diagnosis of fragile X syndrome, type 1 diabetes mellitus and malabsorption syndrome caused by giardias is presented. Fragile X syndrome is a rare entity. This particular association with the other two conditions has become the first case reported in the country. Some of the genetic characteristics of this syndrome are presented as well as the main clinical and diagnostic features. Therapeutic procedures followed with this patient are also stated. It was concluded that fragile X syndrome is one of the most common hereditary disorders and is unknown for most of the population and health professionals. Therefore, it is necessary to spread knowledge on this issue so that families with mentally retarded patients can receive the appropriate genetic counseling. Key words: Fragile x syndrome; diabetes mellitus type I; malabsorption syndromes
Key words: fragile x syndrome, diabetes mellitus type I, malabsorption syndromes.
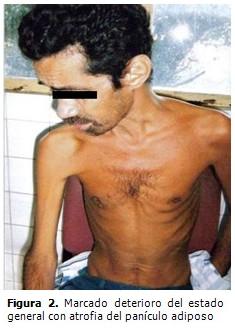
INTRODUCCIÓN El síndrome frágil X (SFX) conocido también como síndrome de Martin Bell o síndrome de Fraxa es la primera causa de retraso mental hereditario. (1) Su nombre se debe a los estudios que, además de descubrir su origen genético, encontraron que personas con ciertas características mentales y físicas, tenían en su cromosoma X un fragmento parcialmente roto. (2) El SFX se presenta tanto en el hombre como en la mujer aunque afecta más intensamente a los varones, (3) por esto las mujeres pueden presentar esta anomalía en cualquiera de los cromosomas sexuales X, mientras que los hombres lo padecen en el único cromosoma sexual X que poseen. (4) A pesar de ser uno de los trastornos hereditarios más frecuentes, resulta desconocido para la población en general y la mayoría de los profesionales relacionados con la salud y la educación, quienes poseen datos parciales e incompletos acerca de esta entidad, por lo que su diagnóstico suele ser tardío y con mucha frecuencia erróneo. (2) Esta enfermedad es la segunda causa genética de retraso mental y la forma más frecuente de retraso mental hereditario, con una frecuencia estimada de 1:2000 varones y explica entre el 4 % y 8 % de todos los retrasos mentales en varones y 1:6000 en mujeres, (5) es también responsable de la mayoría de los casos de trastornos hereditarios en el desarrollo psicomotor. (6) La anomalía se debe a una mutación genética del ADN que afecta tanto a las células sexuales (óvulos y espermatozoides) como a los otros tipos de células del organismo. (2) Este trastorno ocasiona una clase de mutación poco habitual: una secuencia reiterada del código del ADN, llamada repetición de triplete; cuanto mayor sea el número de esta secuencia repetida, más alta es la probabilidad de que la persona sufra alteraciones graves. (7) Esta enfermedad resulta en particular de un defecto en un gen llamado FMRA. (7) El defecto de este gen es una repetición del trinucleótido CGG (Triplete Citosina–Guanina–Guanina) en una parte del mismo que regula su expresión. (8) Ha motivado la realización de este trabajo el haber diagnosticado un paciente con SFX asociado a diabetes mellitus tipo 1 y síndrome de malabsorción secundario a giardiasis lo cual no ha sido reportado aún en Cuba ni en la literatura mundial. PRESENTACIÓN DEL CASO Paciente de 28 años de edad, de sexo masculino, de color de piel blanca, nacido de parto eutócico, en un medio rural; con el antecedente de su abuelo paterno muerto a los 50 años de infarto del miocardio, un hermano fallecido a los 20 años de meningoencefalitis bacteriana, así como una hermana y una tía materna con retardo mental moderado. Refirió el padre que el paciente presentó dificultades en el aprendizaje y trastornos de conducta desde los 8 o 10 años, por lo que abandonó la escuela por retardo mental, habiendo ocurrido lo mismo con el hermano fallecido y la hermana a los 15 años por tener poco aprovechamiento en la escuela. A los 15 años fue ingresado por convulsiones y se le diagnosticó una epilepsia, fue dado de alta del hospital municipal con fenitoína (50mg), 3 tabletas diarias, tratamiento que abandonó 2 años después, hizo además convulsiones ocasionales. Hacía un año había presentado diarreas y fue atendido en varias oportunidades por su médico con mejoría transitoria; las diarreas eran múltiples, más acentuadas por la noche, de color amarillo oro, flotaban en el agua y de olor rancio, 10 días antes de su ingreso comenzó con ansiedad y excitación más acentuadas que de costumbre, acompañándose de polidipsia y poliuria, por lo que acudió a su facultativo el cual le indicó examen de glucemia de urgencia, enviándolo al Hospital Universitario Manuel Ascunce Domenech donde fue hospitalizado para realizar estudio y tratamiento. Examen Físico Presentó caquexia, se encontraba afebril, con deshidratación ligera, la piel suave, aterciopelada, cara alargada, mentón y orejas prominentes. (Figura 1).
Sistema osteomioarticular: presentaba hipotonía, hiperlaxitud e hiperextensibilidad de todas las articulaciones, sobre todo las menores. (Figura 2).
Examen cardio-respiratorio: sin alteraciones. Tensión arterial: 110/70 mm Hg. Frecuencia cardíaca central: 88 lat. / min. Frecuencia respiratoria: 20 resp/min. Abdomen: sin alteraciones.
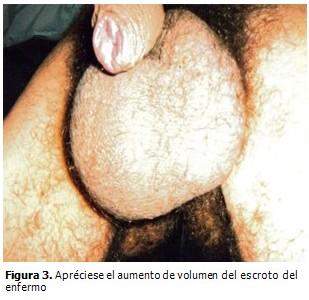
Tacto rectal: normal. Aparato genitourinario: riñones no palpables ni peloteables, fueron todas las maniobras negativas, escroto agrandado de tamaño donde se palpaban ambos testículos aumentados de tamaño. (Figura 3).
Sistema hemolinfopoiético: no presentó adenopatías ni esplenomegalia.
Sistema nervioso: comportamiento autista, retraso mental severo, hiperactividad, ansiedad, trastornos de la atención, habla reiteradamente con ecolalia, no cooperó con el examen, se encontraba desorientado en tiempo, espacio y persona. El examen físico de los pares craneales fue normal excepto disminución de la agudeza visual de ambos ojos. Fondo de ojo: normal.
Estudios analíticos e imagenológicos
Hemoglobina: 10,5 g/dL. Hematocrito: 0,34. Leucocitos: 10,150 x 109/L con fórmula diferencial normal. Velocidad de sedimentación globular: 60 mm/1ª h. Glucemias repetidas: elevadas, la menor con cifras de 15 mmol/L. Enzimas hepáticas, fosfatasa alcalina, LDH, GGT, enzimas pancreáticas, estudio de la función renal, iones, ácido úrico: todos normales. Tiempos de protrombina, coagulación, sangrado: dentro de límites normales. Conteo de ADDIS (2 horas) y otros estudios de sedimento urinario: normales.
Proteínas totales: 5,0 g %. Albúmina: 2,9 g %. Globulina: 2,1 g %. Calcio en sangre: 1,6 mmol/L. Calcio en orina: 1,2 mmol/L. Estudio coprológico: huevos de Ascaris lumbricoides y Giardia lamblia. Coprocultivos: negativos. Prueba de Mantoux: 2 mm.
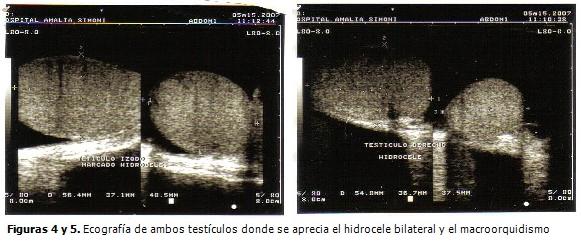
Radiografía de tórax: normal. Ecografía abdominal y de próstata: normal. Ecografía de escroto: hidrocele bilateral, con marcado aumento de ambos testículos. (Figuras 4 y 5).
Tomografía axial computarizada (TAC) de cráneo: atrofia cortical, resto sin alteraciones. Electroencefalograma: signos de irritación cortical. Electrocardiograma: normal. Ecocardiografía: dentro de la normalidad.
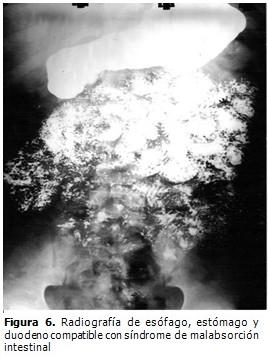
Radiografía de esófago, estómago y duodeno con tránsito intestinal: compatible con síndrome de malabsorción intestinal (Figura 6).
Frotis y biopsia yeyunal: aplanamiento total de las vellosidades compatibles con malabsorción intestinal, de acuerdo con este hallazgo se realizaron estudios de Sudan III en heces fecales: positivo. Amilasa pancreática y lipasa: normales. Colonoscopia: sin alteraciones.
Se impuso tratamiento dietético y con insulina lenta U-100 para la diabetes mellitus tipo I, así como metronidazol (250 mg), 3 tabletas al día para la Giardia lamblia y mebendazol (150mg), 2 tabletas al día por 3 días para el Ascaris lumbricoides, vitaminoterapia y minerales; albumina humana al 20 % e hidratación con solución salina. Para el tratamiento del SFX se empleó metilfenidato y clonidina sin obtener mejoría. Se añadió al tratamiento carbamazepina (200mg), a razón de 15 mg/kg/día en dosis fraccionadas, mejoró de manera considerable su cuadro clínico, otorgándole el alta a los 50 días totalmente recuperado, e indicándole a toda la familia estudios genéticos para detectar otros individuos con estas alteraciones o relacionados ella.
DISCUSIÓN
En el SFX el cariotipo muestra un cromosoma X que presenta una constricción cercana al extremo del brazo largo, esta se suele fragmentar en los preparados y de ahí deriva el término de cromosoma X frágil, esta anomalía cromosómica es después del Síndrome de Down la causa más frecuente de retraso mental. (9) Las mujeres trasmisoras suelen ser normales, pero en ocasiones presentan ligero retraso mental, (9) en la familia del enfermo se encontró una hermana y una tía con anormalidades de moderadas a severas.
Se debe resaltar que las técnicas actuales no permiten el diagnóstico prenatal (9) y se heredan de modo tal, que tanto hombres como mujeres pueden ser portadores de la premutación, (9) los hombres portadores de la premutación la transmiten al 100 % de sus hijos a partir de los cuales puede producirse la mutación completa y tener varones afectados, por esta razón su herencia se conoce como ligada al cromosoma X. (10) Este paciente tuvo un hermano que falleció a los 20 años con características similares, una hermana con retraso mental y algunas características somáticas propias de la enfermedad, e igualmente una tía materna con retraso mental pero sin alteraciones fenotípicas.
Los varones que heredan la mutación completa de sus madres portadoras de la premutación, son principalmente los afectados, sin embargo las hijas de estas mujeres pueden también heredar la mutación completa y presentar retraso mental pero mucho más ligero que sus hermanos, (7, 10) como se pudo apreciar en los familiares de este paciente.
No se pudo realizar el análisis familiar por la lejanía de sus domicilios.
Alrededor del 20 % de los varones son portadores de mutación del cromosoma X frágil y transmiten el rasgo a los nietos afectados a través de sus hijas recibiendo el nombre de: varones transmisores, alrededor del 30 % de las portadoras femeninas están afectadas (es decir, presentan retraso mental). El riesgo de retraso mental en los hermanos de un varón transmisor es del 9 %, mientras que en los nietos varones de los varones transmisores este riesgo se eleva al 4 0 % (9, 10) este riesgo posicional suele denominarse: paradoja de Sherman (11) y pudo ocurrir en este caso.
Las manifestaciones clínicas comienzan en la infancia con retraso psicomotor fundamentalmente del desarrollo del lenguaje y ciertas alteraciones de la conducta y las relaciones sociales, parecidas a las observadas en los niños con autismo, además existen anormalidades del lenguaje que incluyen errores semánticos, ansiedad y ecolalia (1) tal como presentó en el estudio.
Al nacimiento, el peso, talla y circunferencia cefálica son normales o superiores al 75 percentil, (2) como se pudo constatar. Tienen alteraciones craneofaciales características: la cara es alargada, el mentón es grande, las orejas prominentes y muestran alteraciones propias de defectos del tejido conectivo; el paladar es alto, tienen hipotonía, hiperlaxitud ligamentosa, pies planos, escoliosis, prolapso de la mitral y piel fina y aterciopelada, (1-3, 9,10) excepto las alteraciones cardíacas, los pies planos y la escoliosis el enfermo presentaba el resto de estas alteraciones. El 60 % de los pacientes presentan epilepsia y los varones post-puberales macro-orquidismo de gran valor diagnóstico clínico (1, 9, 10) y que fueron encontrados en este paciente. La única característica distintiva que puede detectarse en al menos un 80 % de los varones post-puberales es el macroorquidismo. (10)
El diagnóstico del SFX se realizaba inicialmente gracias a la expresión citogenética de un sitio frágil en X q 27,3 (2, 4) esta prueba no se utiliza en la actualidad, ya que su sensibilidad y especificidad son insuficientes. (11) Actualmente se utiliza la hibridación de Southern o se puede usar la reacción en cadena de la polimerasa (PCR) (12); también se emplea como criterio diagnóstico el puntaje clínico desarrollado por De Vries a partir de 1999, basado en las características más frecuentes señaladas por Hagerman (12) y que son: comportamiento autista, retraso mental, hiperactividad, problemas de atención, aleteo de brazos, trastornos visuales, lenguaje reiterativo, articulaciones hiperextensibles, testículos grandes y orejas prominentes. Con excepción del aleteo de los brazos, este paciente presentó el resto de las alteraciones.
No se ha encontrado en la literatura la asociación de SFX con diabetes mellitus tipo 1, lo cual puede ser un hecho puramente casual; una asociación causal requeriría de observaciones y estudios posteriores. En la familia del paciente no se reportaron enfermos diabéticos.
La giardiasis se caracteriza por diarreas, malabsorción y pérdida de peso (13) como presentó este enfermo. Se debe señalar que la infección se auto-limita, pero es posible que evolucione como una enfermedad indolente con pérdida de peso progresiva, (13) como se pudo constatar en el estudio.
En el tratamiento lo primero a considerar es la prevención, y dentro de esta, la prevención primaria, que incluye un adecuado asesoramiento genético al resto de los miembros de la familia. (9)
En este paciente se utilizó durante 2-3 años la difenilhidantoína, la cual abandonó, presentando crisis convulsivas en múltiples oportunidades. En los niños de edad preescolar se utilizan medicamentos estimulantes como el metilfenidato o medicamentos calmantes como la clonidina, (14) los cuales fueron empleados en el enfermo. Como medicamentos anticonvulsionantes se utilizaron: la carbamazepina, el ácido valproico y la gabapentina. (6, 14) en este caso se empleó la primera, con excelentes resultados.
El paciente es asistido por consulta y después de 6 meses se encuentra libre de diarreas y convulsiones con la diabetes mellitus controlada y un buen estado general.
REFERENCIAS BIBLIOGRÁFICAS
1. Llanio Navarro R, Lantigua Cruz A, Fernández Naranjo A, Batule Batule M, Paniagua Estévez M, Matarama Peñate M. Síndromes. En: Llanio Navarro R, editores. Síndromes pediátricos. La Habana: Editorial de Ciencias Médicas; 2005: p. 428-525
2. Lugones Botell M, Miyar Pieiga E, Ramírez Bermúdez M,Martínez La Fuente AM. Síndrome Frágil X. Rev Cubana Med Gen Integr. 2006;22(3):2-10
3. Neri G, Opitz JM. Sixty years of X-linked mental retardation: a historical footnote. Am J Med Genet Fall. 2000;97(3):228-33
4. Jin P, Warren ST. Understanding the molecular basis of fragile X syndrome. Hum Mol Genet. 2000;9:901-8
5. De Smedt B, Swillen A, Verschaffel L, Ghesquiere P. Mathematical learning disabilities in children with 22q11.2 deletion syndrome: a review. Dev Disabil Res Rev. 2009;15(1):4-10
6. Ferrando LM, Banús Gómez P, López Pérez G. Aspectos cognitivos en niñas con síndrome X frágil. Rev Neurol. 2004;38 Suppl 1:53-7
7. Bailey DB, Raspa M, Bishop E, Holiday D. No change in the age of diagnosis for fragile X syndrome: findings from a national parent survey. Pediatrics. 2009;124(2):527-33
8. Biancalana V, Toft M, Le Ber I. FMR1 premutations associated with fragile X-associated tremor/ataxia syndrome in multiple system atrophy. Arch Neurol. 2005;62(6):962-6
9. Hersh JH, Saul RA. Committee on genetics. Clinical Report—Health Supervision for Children With Fragile X Syndrome. Pediatrics. 2011;127(5):994
10. Hall SS, Lightbody AA, Huffman LC, Lazzeroni LC, Reiss AL. Physiological correlates of social avoidance behavior in children and adolescents with fragile x syndrome. J Am Acad Child Adolesc Psychiatry. 2009;48(3):320-9
11. Garber KB, Visootsak J, Warren ST. Fragile X syndrome. Eur J Hum Genet. 2008;16(6):666-72
12. Reiss A, Dant C. The behavioural neurogenetics of fragile X syndrome: analyzing gene–brain behaviour relationships in child develop mental psychopathologies. Dev Psychopathol. 2003;15:927-68
13. Sears CL. Giardiasis. En: Arend WP, Armitage JO, Clemmons DR, Drazen JM, Griggs RC, LaRusso N, editores. Cecil Medicine. Philadelphia: Saunders Elsevier; 2008: p. 35-54
14. Hagerman RJ, Berry-Kravis E, Kaufmann WE, Ono MY, Tartaglia N, Lachiewicz A, et al. Advances in the treatment of fragile X syndrome. Pediatrics. 2009;123(1):378-90
Recibido: 22 de febrero de 2011.
Aprobado: 12 de diciembre de 2011.
Rafael Pila Pérez. Especialista de II Grado en Medicina Interna. Profesor Titular. Hospital Universitario Manuel Ascunce Domenech. Camagüey. Correo electrónico: vadolfo@finlay.cmw.sld.cu


{kind=link}