










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkMediSur
versión On-line ISSN 1727-897X
Medisur vol.11 no.5 Cienfuegos oct. 2013
PRESENTACIÓN DE CASO
Síndrome de Proteo. Presentación de un caso
Proteus Syndrome. A Case Report
Edelmis Pérez SalomónI , Noralis ConcepciónII , Mayris Terrero MoraII , Zaida Rodríguez NavarroII , Alexander Torres MolinaI
I Hospital Pediátrico Pedro Soto Alba, Moa, Holguín, Cuba
II Policlínica Universitaria Rolando Monterrey, Moa, Holguín, Cuba
RESUMEN
El síndrome de Proteo es un raro y complejo trastorno genético caracterizado por aparición esporádica, distribución en mosaico y evolución progresiva de lesiones hamartomatosas, que afectan la mayoría de los tejidos de origen mesodérmico. El diagnóstico es clínico y se establece según criterios bien establecidos. Se presenta un caso típico en un escolar de ocho años de edad con manifestaciones clínicas compatibles con el diagnóstico de este síndrome. Se realizó estudio y descripción clínica del hábito externo; se destacaron como principales rasgos distintivos: hemihipertrofia progresiva de región glútea, pierna y pie izquierdo, hemangioma plano y dilatación de venas hipogástricas en abdomen, escoliosis dorso-lumbar y cifosis dorsal marcada, aumento del tejido graso en el dorso, aspecto cerebriforme de la piel de la planta del pie izquierdo e hiperostosis mastoidea. Por lo poco común de esta entidad se decidió la presentación del caso.
Palabras clave: síndrome de Proteo, niño, informes de casos.
ABSTRACT
Proteus syndrome is a rare and complex genetic disorder characterized by sporadic occurrence, mosaic distribution and gradual evolution of hamartomatous lesions, affecting most of the mesodermal tissues. Diagnosis is performed on clinical basis and according to well-established criteria. A typical case of an 8-year-old schoolchild with clinical manifestations consistent with the diagnosis of this syndrome is presented. A clinical study and description of the body habitus was conducted. The main distinctive features were: progressive hemihypertrophy of the gluteal region, left leg and foot, flat hemangioma and dilatation of hypogastric veins in the abdomen, dorso-lumbar scoliosis and marked dorsal kyphosis, increased adipose tissue in the back, cerebriform aspect of the skin of the sole of the left foot as well as hyperostosis of the mastoid bone. This case is presented due to the rarity of the entity.
Key words: Proteus syndrome, child, case reports.
INTRODUCCIÓN
El síndrome de Proteo (SP) fue descrito como entidad nosológica por primera vez en 1979 por Cohen y Hayden, sin embargo no recibió la denominación hasta 1983 en un artículo publicado por Wiedemann et al, para describir cuatro niños con malformaciones hamartomatosas congénitas.1 Wiedemann utilizó como referencia, para abordar las alteraciones estructurales que definen el síndrome, a un legendario héroe griego, transformado por los dioses en un demonio inmortal del mar, el cual podía cambiar de forma a voluntad, y que fue descrito en La Odisea por Homero.2 Es considerado un raro desorden genético que se produce por mutación en el gen 10q23.31, encargado de activar de forma esporádica el crecimiento de varios tejidos (epidérmico, conectivo, óseo, adiposo, endotelial) durante el periodo de desarrollo embrionario, por lo que se manifiesta generalmente al nacimiento o en los primeros años de vida y se caracteriza por gigantismo parcial de manos y pies, nevos pigmentados, hemihipertrofia cutánea, tumores hamartomatosos subcutáneos, macrocefalia y anomalías craneales.3-5 Desde su primera descripción hasta la actualidad solo se han diagnosticado poco más de 500 casos en países desarrollados, con afectación por igual de ambos sexos. Constituye una enfermedad progresiva que conlleva a desfiguraciones severas, limitaciones físicas y reducción de la calidad de vida.6-8 No se encontraron referencias en las publicaciones médicas cubanas donde se describa la existencia de pacientes con esta extraordinaria entidad clínica por lo que el objetivo de este trabajo es presentar un caso diagnosticado con el síndrome de Proteo en Moa, provincia Holguín.
PRESENTACIÓN DEL CASO
Escolar de sexo masculino de ocho años de edad, color de piel negra, procedencia urbana, nacido de parto distócico por cesárea, pesó al nacer 2 830 gramos, apgar 9/9, sin antecedentes patológicos prenatales o familiares de interés. Al nacimiento se presentó hemihipertrofia de la región glútea, pierna y pie izquierdo, con macrodactilia, displasia del desarrollo de cadera, genusvalgo bilateral y hemangioma plano en abdomen y espalda. A los dos años de edad se detectó escoliosis dorso-lumbar y cifosis dorsal pronunciada, hiperplasia de la musculatura de la región glútea y muslo izquierdo con aumento de tejido celular subcutáneo y tejido adiposo a nivel dorsal. Tres años después fue necesario realizar extirpación quirúrgica de la falange distal del segundo dedo del pie izquierdo por hipertrofia cortical marcada que le imposibilitaba ponerse zapato. Todas las malformaciones anteriores se pueden observar en las siguientes figuras. (Figuras 1,2,3).
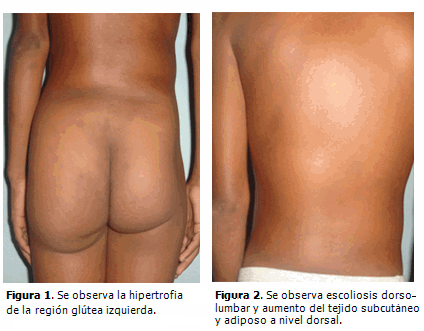
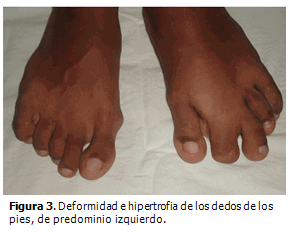
En la actualidad es evidente la progresión de la enfermedad. Presenta deformidad de los huesos del metatarso y de las falanges de los dedos del pie izquierdo así como aspecto cerebriforme de la piel que cubre la planta del pie e hiperostosis de las mastoides. (Figura 4).

Otros datos de interés fueron la aparición de máculas hipercrómicas con bordes bien definidos en el dorso y dilatación venosa en la región hipogástrica de la pared abdominal.
DISCUSIÓN
Se han postulado diversas propuestas de criterios diagnósticos para el síndrome de Proteo, las cuales se basan fundamentalmente en parámetros clínicos. Los primeros fueron propuestos por Wiedemann y colaboradores, en 1983, en una serie inicial de cuatro pacientes,2 aunque ninguno de ellos presentaba la totalidad de la sintomatología descrita. Costa y colaboradores, en 1985,9 y Clark,10 en 1987, dividen los signos clínicos del SP, según su frecuencia, en principales, frecuentes y ocasionales. Posteriormente, Samlaska et al,11 en 1989, con una serie de 34 pacientes y Sayama et al,12 en 1994, en una revisión de 63 casos, proponen una relación según los hallazgos clínicos más frecuentes. Para estos autores los criterios frecuentes eran los encontrados en más de la mitad de los pacientes. Hotamisligil,13 en 1990, fue más estricto dando una puntuación a cada sintomatología. Así, otorga a la macrodactilia y/o hemihipertrofia, cinco puntos; al engrosamiento palmar o plantar, cuatro puntos; los tumores subcutáneos y lipomas, cuatro puntos; los nevos epidérmicos, tres puntos; la macrocefalia o las exostosis craneales 2,5 puntos y al resto de anormalidades menores un punto. El diagnóstico se establecía si el paciente sumaba 13 o más puntos. Para clarificar todos estos criterios, en marzo de 1998, se convocó la primera conferencia nacional del SP para padres y familiares (National Institutes of Health de Bethesda, Maryland, EE.UU.), donde se establecieron una serie de criterios diagnósticos, recomendaciones, guías de evaluación y diagnósticos diferenciales.14 Como criterios generales (obligatorios) se incluyeron la distribución de las lesiones en mosaico, el curso progresivo y la aparición esporádica, mientras que los criterios específicos se clasificaron en tres categorías A, B y C. (Tabla 1). El criterio incluido en la categoría A, en presencia de todos los criterios obligatorios es suficiente para el diagnóstico, así como dos de la categoría B o tres de la C.14 Como diagnósticos diferenciales es necesario destacar que se descartaron otras enfermedades hamartomatosas como la neurofibromatosis, los síndromes de Kipplel-Trenaunay, Mafucci, Bannayan-Riley-Ruvalcaba, Parkes-Weber, del nevo epidérmico, la lipomatosis simétrica y lipomatosis encefalocraneocutánea.1-4 La mayoría de los casos de síndrome de Proteo, se clasificaban como neurofibromatosis de Von Recklinghausen hasta que se establecieron los criterios diagnósticos de los dos tipos de neurofibromatosis en 1988. Sólo dos años antes, los antropólogos Tibles y Cohen habían identificado el síndrome de Proteus como el trastorno que padeció Joseph Merrick, "el hombre elefante", considerado hasta entonces como un caso de neurofibromatosis.1,7
El paciente que se presenta en este trabajo reúne los tres criterios obligatorios así como dos de los criterios específicos incluidos en la categoría B (nevus epidérmicos, crecimiento desproporcionado de miembros e hiperostosis mastoidea) y dos de la categoría C (tejido adiposo disrregulado y malformaciones vasculares) por lo que se puede concluir que padece de síndrome de Proteus.
REFERENCIAS BIBLIOGRÁFICAS
1. Guerra Tapia A, Rodríguez Vázquez M. Síndrome de Proteo. Piel. 2001;16(5):248-52
2. Wiedemann HR, Burgio GR, Aldenhoff P, Kunze J, Kaufmann HJ, Schirg E. The proteus syndrome. Eur J Pediatr. 1983;140:5-12
3. Redondo P. Malformaciones vasculares (I). Concepto, clasificación, fisiopatogenia y manifestaciones clínicas. Actas Dermosifiliogr. 2007;98(3):141-58
4. Schnake Ch, Vejar L, Solar M, Ibanez R, Lacassie Y. Síndrome Proteus: Contribución a su delineación clínica. Rev Chil Pediatr. 1986;57(6):585-94
5. Sánchez López M, Martínez Fernández R, Santamaría Carro A. Manifestaciones oculares en el síndrome de Proteus. Arch Soc Esp Oftalmol. 2007;82(3):175-8
6. Capurro NJ, Carignano AM, Ottino A. Proliferación fibrosa cerebriforme, correlación con el síndrome de Proteus. Patología. 2008;46(4):351-4
7. Fernández Mayoralas M, Fernández Jaén A, Calleja Pérez B, Muñoz Jareño N. Enfermedades neurocutáneas. JANO [revista en Internet]. 2007 [citado 22 Oct 2011];1:[aprox. 6p]. Disponible en: http://www.jano.es/ficheros/sumarios/1/0/1667/87/00870091-LR.pdf
8. Bennàsar A, Ferrando J. Síndromes de neoplasias múltiples familiares con manifestaciones cutáneas. Med Cutan Iber Lat Am. 2009;37(2):71-8
9. Costa T, Fitch N, Azouz EM. Proteus syndrome: report of two cases with pelvic lipomatosis. Pediatrics. 1985;76(6):984-9
10. Clark RD, Donnai D, Rogers J, Cooper J, Baraitser M. Proteus syndrome: an expended phenotype. Am J Med Genet. 1987;27(1):99-117
11. Samlaska CP, Levin SW, James WD, Benson PM, Walker JC, Perlik PC. Proteus syndrome. Arch Dermatol. 1989;125(8):1109-14
12. Sayama K, Hato N, Matsuda O, Shiraishi S, Miki Y. Proteus syndrome. Dermatology. 1994;189(4):392-5
13. Hotamisligil GS. Proteus syndrome and hamartoses with overgrowth. Dysmorphol Clin Genet. 1990;4:87-102
14. Biesecker LG, Happle R, Mulliken JB, Weksberg R, Graham JM, Viljoen DL, et al. Proteus syndrome: diagnostic criteria, differential diagnosis, and patient evaluation. Am J Med Genet. 1999;84(5):389-95
Recibido: 19 de noviembre de 2011.
Aprobado: 04 de octubre de 2013.
Edelmis Pérez Salomón. Especialista de I Grado en Pediatría. MSc. en Atención Integral al Niño. Profesor Asistente. Hospital Pediátrico Pedro Soto Alba. Moa. Holguín. Correo electrónico: edelmis@moa.hlg.sld.cu


{kind=link}