










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkINTRODUCCIÓN
El síndrome de Baller-Gerold fue descrito por primera vez en 1950 por F. Baller, posteriormente en 1959, M. Gerold completa su descripción. 1,2,3
Se sabe que la mutación responsable del síndrome se localiza en el gen RECQL4, mutación encargada de la síntesis de una proteína que tiene una función aún no bien comprendida, sin embargo, los investigadores creen que esta proteína ayuda a estabilizar la información genética en las células del cuerpo. Es probable que también el gen esté relacionado con el proceso de reparación y replicación del ADN. 4,5,6
El síndrome de Baller-Gerold está considerado como una enfermedad hereditaria, muy rara, caracterizada por craneosinostosis, cúbito corto y curvado y aplasia o hipoplasia del radio que no suele ser simétrica, retraso del crecimiento, retraso del desarrollo psicomotor y diferentes malformaciones craneofaciales, cardíacas, renales y esqueléticas. 1,4,5
Se puede sugerir un diagnóstico prenatal, por análisis de las vellosidades coriónicas, cuando se han identificado mutaciones patogénicas del gen RECQL4 en el caso índice (homocigoto o heterocigoto). La ecografía se puede utilizar para detectar las anomalías de miembros o una forma anormal del cráneo. 7,8,9,10
Durante los primeros meses de vida puede aparecer una poiquilodermia inconstante. El retraso en el crecimiento está casi siempre presente, habitualmente alrededor de 4 desviaciones estándar. (2,8 10)
También se puede observar aplasia o hipoplasia durante la infancia. La inteligencia es por lo general normal. Los pacientes tienen una predisposición al desarrollo de patologías malignas, en particular, de osteosarcoma. 7,10
Los principales diagnósticos diferenciales incluyen los síndromes de Rothmund-Thomson (RTS) y RAPADILINO (por sus siglas en inglés), también secundarios a mutaciones del gen RECQL4. Diversos autores han sugerido un continuum fenotípico entre estas entidades, es posible que estas enfermedades representen diferentes expresiones de una misma patología. 10
Otros diagnósticos diferenciales incluyen el síndrome de Roberts (SR) y la anemia de Fanconi, habitualmente asociados a anomalías del eje radial, pero no a craneosinostosis y el síndrome de Saethre-Chotzen, caracterizado por craneosinostosis coronal, habitualmente sin anomalías del eje radial. 10
El tratamiento consiste en cirugía de la craneosinostosis bilateral durante los primeros 6 meses de vida y, en el caso que sea necesario, pulgarización del dedo índice para la reconstrucción del pulgar. 10
Se presenta el caso por su baja incidencia y prevalencia por lo que es considerada una enfermedad rara.
PRESENTACIÓN DEL CASO
Se presenta el caso de una recién nacida, de color de piel blanca, hija de madre de 21 años, con antecedentes patológicos personales de salud aparentes. Esta paciente nació producto de parto distócico por cesárea a las 35,5 semanas, con diagnóstico prenatal de restricción del crecimiento intrauterino tipo III e hipoplasia del cuerpo calloso. Tenía como peso al nacer, 1240 gramos. Según la evaluación nutricional presentó retraso de crecimiento intrauterino (RCIU) por debajo del 3er percentil, Apgar 5-7, se necesitó realizar reanimación en sala de partos y ventilación mecánica no invasiva modalidad: presión positiva continua en la vía aérea (CPAP) (por sus siglas en inglés).
La paciente fue transferida al Servicio de Neonatología del Hospital General Universitario Dr. Gustavo Aldereguía Lima. Al realizar estudios de diagnóstico por imágenes se constataron múltiples malformaciones músculo-esqueléticas, aplasia radial, pulgares aplásicos, malformaciones de la parrilla costal, clinodactilia de todos los dedos de miembros superiores, antebrazos hipoplásicos, clinodactilia del miembro inferior izquierdo. (Fig 1, Fig.2 y Fig.3).
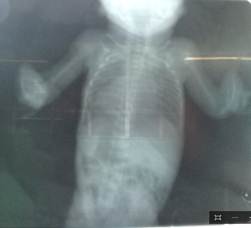
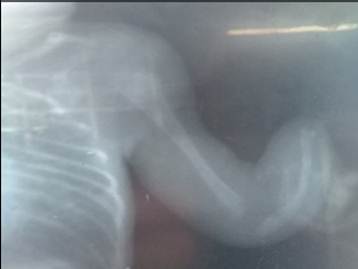
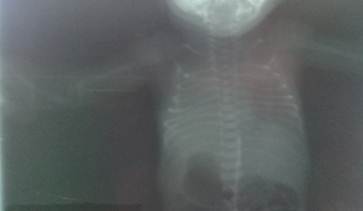
La paciente presentaba malformaciones cráneo faciales: frente hacia atrás, cabeza en forma alargada y cónica, mandíbula anormalmente pequeña, orejas malformadas y con implantación baja y paladar ojival.
Se diagnosticaron en este caso, anomalías del sistema nervioso central (SNC) del tipo hipoplasia cerebelosa, mega cisterna magna y disgenesia del cuerpo calloso. A la paciente se le realizó alimentación parenteral por varios días, fue imposible establecer vía oral adecuada por ausencia y/o incoordinación de los reflejos de succión-deglución como expresión de sus malformaciones a nivel de SNC.
La paciente evolucionó con complicaciones asociadas a su nacimiento temprano y la restricción del crecimiento, se procedió a su egreso con más de 100 días de vida, del Servicio de Neonatología del Hospital General Universitario Dr. Gustavo Aldereguía Lima de Cienfuegos para su seguimiento y posterior manejo ambulatorio.
DISCUSIÓN
El síndrome de Baller-Gerold clínicamente se caracteriza por anomalías cráneo faciales: craneosinostosis, frente hacia atrás, oxicefalia, micrognatia, prognatismo mandibular y hemangioma capilar, orejas malformadas y situadas más abajo de lo habitual, hipertelorismo, puente nasal prominente, epicantus y filtrum amplio, fisura palatina, úvula bífida y paladar ojival; anomalías esqueléticas fundamentalmente de los miembros superiores: aplasia radial o cubital y huesos carpianos ausentes o fusionados, pulgares aplásicos o hipoplásicos y frecuentes malformaciones vertebrales, anomalías del sistema nervioso: polimicrogiria e hidrocefalia y anomalías del sistema cardiovascular: malformaciones cardíacas tales como: comunicación interventricular y estenosis valvular subáortica y además anomalías del sistema urogenital: malformaciones renales. 1,8,9,10
El diagnóstico es básicamente clínico, especialmente basado en las siguientes manifestaciones: craneosinostosis coronaria, defectos en los dedos y retraso del crecimiento 10) coincidente con el caso descrito.
La prueba de genética molecular confirmó el diagnóstico, la paciente mostró una mutación en el gen RECQL4, único gen conocido hasta la fecha vinculado a este síndrome. 10

