










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Medicina
versión On-line ISSN 1561-302X
Rev cubana med v.41 n.4 Ciudad de la Habana jul.-ago. 2002
Presentación de casos
Centro Internacional de Restauración Neurológica
Homocistinuria clásica. Presentación de 1 caso
Dr. Liván Rodríguez Mutuberría,1 Dr. Reynaldo Galvizu Sánchez2 y Dr. Eduardo Álvarez González2
Resumen
Se presentó un paciente joven masculino que ingresó en el Centro Internacional de Restauración Neurológica por retraso mental ligero y epilepsia. Se evaluó minuciosamente y se encontró además, un hábito marfanoide caracterizado por dolicostenomelia, aracnodactilia, pectus carinatum, escápula alada y pie plano bilateral. Se consideró como probables diagnósticos, el síndrome de Marfán, el de X frágil y la homocistinuria clásica. Los dos primeros se excluyeron de acuerdo con los datos clínicos y el resultado de los estudios complementarios. Se concluyó como una homocistinuria clásica, teniendo en cuenta la asociación de retraso mental ligero, epilepsia, osteoporosis y hábito marfanoide. Finalmente, el diagnóstico estuvo apoyado por la prueba de cianuro-nitroprusiato en orina que resultó positiva. Se hizo una breve revisión de la base patogénica de la enfermedad.
DeCS: HOMOCISTINURIA/diagnóstico; EPILEPSIA; SINDROME DE MARFAN; RETARDO MENTAL; NITROPRUSIATO; CIANUROS.
El término homocistinuria corresponde a una anomalía bioquímica relacionada con el metabolismo de los aminoácidos (aas); su forma más común, y la de mayor importancia en el adulto, es la deficiencia de cistationina beta sintetasa, cuya expresión clínica está conformada por retraso mental, hábito marfanoide y fenómenos tromboembólicos.1
En la literatura se han reportado hasta 300 casos bien comprobados. Los estudios de búsqueda en neonatos han conducido a una frecuencia, de 1 por cada 200 000 nacimientos en Estados Unidos, lo que establece la baja frecuencia con que se presenta esta entidad en la práctica clínica diaria, aunque países como Irlanda han reportado frecuencias más elevadas, de 1 por cada 40 000 nacimientos.2,3
La enzima afectada es dependiente del fosfato de piridoxal cuyo gen se sitúa en la región q del cromosoma 21 y su función es facilitar la unión de homocisteína y serina, para formar cistationina, por lo que ante un déficit enzimático se acumulan los primeros, disminuyendo la cistationina, su derivado cisteína y la forma disulfuro de ésta, la cistina. El grado de actividad enzimática y, a su vez, la intensidad de las manifestaciones clínicas dependerá de que se trate de una forma homocigótica o heterocigótica.4
Presentación del caso
Paciente masculino de 18 años de edad, sin antecedentes prenatales, perinatales y posnatales de interés. Presentó retardo en el desarrollo psicomotor. A los 8 meses desarrolló una crisis convulsiva, del tipo tónico clónica generalizada en asociación a un cuadro febril por lo que fue interpretado en aquel entonces como una convulsión febril; no necesitó tratamiento con anticonvulsivantes. A los 2 años de edad y sin asociación a fiebre, desarrolló la segunda crisis convulsiva con igual característica que la primera. Con posterioridad, ha desarrollado crisis similares, algunas relacionadas con ajustes en el tratamiento anticonvulsivante. Actualmente se encuentra controlado. Existe retraso escolar manifiesto. Su conducta es desinhibida, algo pueril, fóbica frente a situaciones estresantes. Dentro de los antecedentes familiares de interés existe un tío paterno miope y un tío materno con retraso mental y trastornos de conducta. El examen físico de este paciente arrojó un hábito marfanoide caracterizado por extremidades largas desproporcionadas en comparación con el tronco (dolicostenomelia) (fig. 1), aracnodactilia (fig. 2), pectus carinatum y escápula alada (fig. 3) pie plano bilateral y escoliosis dorsal. Encontramos, igualmente, paladar ojival y resultó positivo el signo de Steinberg y el de Walker - Murdoch. No se pudo demostrar ectopia lentis, pero comprobamos la existencia de miopía ocular. El resto del examen físico resultó negativo.
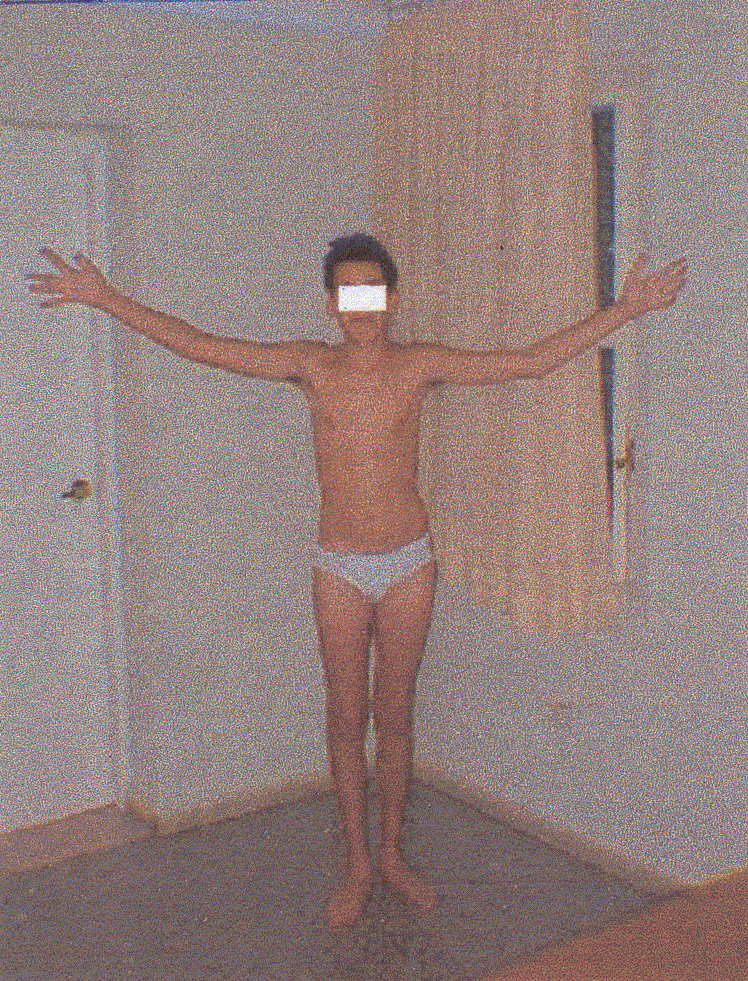
FIG. 1. Dolicoestenomelia.

FIG. 2. Aracnodactilia.

FIG. 3. Escápula alada y pectus carinatum.
Exámenes complementarios
Se realizaron estudios de química sanguínea, hemograma, eritrosedimentación, lipidograma y glucemia, todos los resultados estuvieron dentro de parámetros normales. El electroencefalograma arrojó un trastorno de carácter epileptiforme focalizado en región frontotemporal de ambos hemisferios cerebrales. Según los estudios imagenológicos realizados se comprobó la existencia de osteoporosis en ambas tibias y la pelvis, y mediante densitometría ósea, signos de osteopenia en huesos largos y columna. El ecocardiograma resultó negativo. Le fue aplicado al paciente un test de WAIS con el cual se obtuvo el diagnóstico de débil mental, ubicado en el rango de retraso mental ligero. Finalmente se realizó la prueba de cianuro . nitroprusiato en orina que resultó positiva.
Comentarios
En un principio, valoramos como diagnósticos a tener en cuenta en el paciente expuesto, los siguientes: síndrome de Marfán, síndrome de X frágil y la homocistinuria clásica. El primero fue excluido pues no encontramos daño del aparato valvular del corazón y la asociación de este síndrome con el retraso mental y la epilepsia no está bien documentado en la literatura.5 El síndrome de X frágil, igualmente fue excluido porque suele asociarse a un retraso mental severo y no ligero como el de nuestro paciente y la presencia de macroquídea es frecuente y no la detectamos en el caso analizado.6
Consideramos la homocistinuria clásica como el diagnóstico definitivo ya que como entidad nosológica, se caracteriza por la asociación de hábito marfanoide, retraso mental ligero y epilepsia, elementos encontrados en este paciente.7
En cuanto al hábito externo, no solamente suelen encontrarse en estos enfermos las deformidades esqueléticas descritas con anterioridad, si no que pueden venir acompañadas de fracturas patológicas relacionadas con la osteoporosis de huesos largos y columna.7,8 En el paciente no se reportan fracturas, pero se comprobó mediante estudios complementarios la existencia de descalcificación ósea, lo que a la edad de este paciente sólo puede explicarse por la entidad planteada. El retraso mental es un hecho presente hasta en el 80 % de los casos donde incluimos nuestro paciente, alrededor de 20 % de ellos muestran inteligencia promedio o por arriba del promedio.9 La epilepsia, generalmente en forma de convulsiones tónico clónicas generalizadas, se presenta tan sólo entre el 10 y el 15 % asociado a paroxismos en el electroencefalograma, como ocurre en este caso.9 Sin duda alguna, es el fenómeno tromboembólico el que pone en peligro la vida de los enfermos. Pueden verse afectadas venas y arterias de grande y pequeño calibre. La muerte súbita por oclusión coronaria se presenta en el adulto joven con relativa frecuencia, al igual que los accidentes trombóticos cerebrales o renales que conducen a la muerte del 25 % de los enfermos antes de los 30 años.9 No se encontró esta complicación en el paciente.
Fue útil para establecer el diagnóstico, apoyado en la semiología del caso expuesto, detectar el aumento de productos sulfhidrilo mediante la prueba de cianuro - nitroprusiato, que resultó positiva.10
Summary
A male young patient that was admitted at the International Center of Neurological Restoration due to mild mental retardation and epilepsy was presented. He was thoroughly evaluated and it was also found a marfanoid habit characterized by dolichostenomelia, arachnodactyly, pectus carinatum, winged scapula and bilateral flat foot. Marfan's syndrome, the fragile X syndrome and the classic homocystinuria were considered as probable diagnoses. The first two were excluded according to the clinical data and the result of the complementary studies. It was concluded as a classic homocystinuria, taking into account the association of mild mental retardation, epilepsy, osteoporosis and marfanoid habit. Finally, the diagnosis was supported by the cyanide-nitroprusside test in urine that was positive. A brief review of the pathogenic basis of the disease was made.
Subject headings: HOMOCYSTINURIA/diagnosis; EPILEPSY; MARFAN SYNDROME; MENTAL RETARDATION; NITROPRUSSIDE; CYANIDES.
Referencias bibliográficas
- Abbot MH, Folstein SE, Abbey H, Pyeritz RE. Psychiatric manifestations of homocystinuria due to cystathionine beta-synthase deficiency: prevalence, natural history, and relationship to neurologic impairment and vitamin B(6)-responsiveness. Am J Med Genet 1997;26:959-69.
- Franken DG, Boers GHJ, Blom HJ, Cruysberg JRM, Trijbels FJM, Hamel BCJ. Prevalence of familial mild hyperhomocysteinemia. Atherosclerosis 1996;125:71-80.
- Gallagher PM, Ward P, Tan S, Naughten E, Kraus JP, Sellar GC, et al. High frequency (71 %) of cystathionine beta-synthase mutation G307S in Irish homocystinuria patients. Hum Mutat 1995;6:177-80.
- Kim CE, Gallagher PN, Guttormsen AB, Refsum H, Ueland PM, Ose L, et al. Functional modeling of vitamin responsiveness in yeast: a common pyridoxine-responsive cystathionine beta-synthase mutation in homocystinuria. Hum Molec Genet 1997;6:2213-21.
- Aburawi EH, O'Sullivan J, Hasan A. Marfan's syndrome: a review. Hosp Med 2001;62(3):153-7.
- Haddad R, Uwaydat S, Dakroub R, Traboulsi EI. Confirmation of the autosomal recessive syndrome of ectopia lentis and distinctive craniofacial appearance. Am J Med Genet 2001;99(3):185-9.
- Le Parc JM, Molcard S, Tubach F. Bone mineral density in Marfan syndrome. Rheumatology (Oxford) 2001;40(3):358-9.
- Mudd SH, Skovby F, Levy HL, Pettigrew KD, Wilcken B, Pyeritz RE, et al. The natural history of homocystinuria due to cystathionine beta-synthase deficiency. Am J Hum Genet 1995;37:1-31.
- Torre Delgadillo A, Tellez Zenteno JF, Morales Buenrrostro LE. Hyperhomocysteinaemia: physiopathology and medical implications. Rev Invest Clin 2000;52(5):557-64.
- Yoshida Y, Nakano A, Hamada R, Kamitsuchibashi H, Yamamoto K, Akagi H, et al. Patients with homocystinuria: high metal concentrations in hair, blood and urine. Acta Neurol Scand 1992;86(5):490-5.
Recibido: 28 de mayo de 2001. Aprobado: 7 de agosto de 2001.
Dr. Liván Rodríguez Mutuberría. Centro Internacional de Restauración Neurológica, Avenida 25 No. 15805, entre 158 y 160, Cubanacán, Playa, Ciudad de La Habana, Cuba. Correo electrónico: livan@neuro.sld.cu
1 Especialista de I Grado en Medicina.
2 Especialista de I Grado en Neurología.

