










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La enfermedad poliquística renal autosómica dominante es considerada la enfermedad renal genética más frecuente y es la cuarta causa de la enfermedad renal crónica a nivel mundial.1 Afecta a cerca de 1 por cada 1000 nacidos vivos. La hipertensión arterial de inicio temprano y el fracaso de forma progresiva de la función renal, producto del agrandamiento masivo de los quistes, caracterizan la enfermedad.2
Se asocia a la formación de quistes a nivel de otros órganos, principalmente en el hígado y el páncreas, y con anomalías cardiovasculares como los aneurismas arteriales intracraneales y las disecciones vasculares.2 La enfermedad poliquística hepática definida como la presencia de quistes hepáticos de cualquier tamaño, constituye la manifestación extrarrenal más frecuente de la enfermedad poliquística renal autosómica dominante.3 Se describe también la presencia de quistes a nivel de otros órganos abdominales como el páncreas (9 %) y las vesículas seminales (43 %), los cuales cursan de forma asintomática. Los quistes aracnoideos suelen presentarse en el 8 % de los casos. Éstos pueden llevar a la aparición de hematoma subdural.4,5 Dentro de las alteraciones cardiovasculares se describen los aneurismas arteriales intracraneales y las anomalías de las válvulas cardíacas. El prolapso de la válvula mitral es la anomalía valvular que se presenta con mayor frecuencia, entre un 25-40 %. Otras alteraciones cardiovasculares que se pueden presentar son la dilatación de la raíz de la aorta y el aneurisma de la aorta abdominal.6,7
Se describe también la aparición de hernias de la pared abdominal, originadas por la compresión crónica que ejercen los quistes renales y hepáticos, cuando se produce un crecimiento excesivo de estos. Reportes de series de casos han sugerido que las hernias de localización periumbilical e inguinal son las que con mayor frecuencia se presentan, entre un 15-45 % de los pacientes con enfermedad poliquística renal autosómica dominante.8
Los quistes hepáticos se desarrollan un tiempo después que los quistes renales y tienen una proporción de crecimiento anual de aproximadamente 0,9-3,2 %, presentan un curso benigno, que puede existir una ligera elevación de las pruebas de función hepática con el incremento en el número de quistes, pero usualmente no existe una evolución hacia la cronicidad. La cirrosis hepática es un hallazgo tardío en esta enfermedad y cuando se presenta es más común en el anciano. La presencia de ascitis es rara y cuando aparece suele ser en estadios avanzados de la enfermedad. La descompensación de la enfermedad con falla hepática como causa de muerte es rara en la enfermedad poliquística hepática.9
Presentación de caso
Paciente masculino de 60 años de raza blanca con antecedentes patológicos personales de la enfermedad poliquística renal diagnosticada hace 40 años, en seguimiento por nefrología y sin criterio de hemodiálisis. Antecedentes patológicos familiares de padre, hermano e hija con la misma enfermedad, fallece por complicaciones de ésta el padre y el hermano. Dentro de sus hábitos tóxicos destacan el tabaquismo por más de 40 años y el consumo de bebidas alcohólicas de forma ocasional. Ingresa por presentar aumento de volumen del perímetro abdominal y edema en ambos miembros inferiores de 3 meses de evolución. Niega síntomas como fiebre o pérdida de peso.
Al examen físico: paciente orientado en tiempo, espacio y persona con habla coherente. Presencia de ligero tinte ictérico en piel y mucosas. Edema en ambos miembros inferiores hasta la rodilla, blando, no doloroso, con fóvea, normotérmico. Eritema palmar bilateral. Abdomen difícil de explorar por gran distensión, con presencia de red de circulación venosa colateral, percusión timpánica generalizada y maniobra de Tarral positiva, además, hernia a nivel umbilical no dolorosa, con cambios necróticos en el centro (fig. 1).
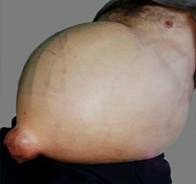
Fig. 1 Abdomen distendido con presencia de red de circulación venosa colateral y hernia umbilical con área de necrosis central.
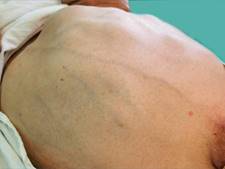
Para mejorar la distensión abdominal del paciente y tomar muestra del líquido ascítico se realizó paracentesis. Se obtuvo 3 litros de líquido claro, cetrino, inodoro, se le realizaron estudios microbiológicos (bacteriológico y BAAR), citoquímico y citológico (fig. 2).

Posterior a la realización del proceder se dejan ver múltiples tumoraciones irregulares que hacen relieve y que ocupan prácticamente toda la cavidad abdominal (fig. 3).
Dentro de los estudios de laboratorio clínico realizados se reporta hemoglobina normal (12,3 g/L), leucocitos normales (8,9 ( 109/L), plaquetas normales (192 ( 109/L), glicemia normal (4,6 mmol/L), creatinina ligeramente elevada (135 umol/L), aspartato aminotransferasa elevada (68 U/L), proteínas totales bajas (51 g/L), albúmina baja (24 g/L), colesterol disminuido (1,5 mmol/L), triglicéridos disminuidos (0,8 mmol/L), bilirrubina total elevada (50 mmol/L) y prolongación del tiempo de protrombina (control 14 segundos, paciente 25 segundos). Electrolitos séricos normales (sodio 137 mmol/L, potasio 4,0 mmol/L). Anticuerpo contra virus C negativo, antígeno de superficie negativo.
Se reporta el citoquímico del líquido ascítico con leucocitosis a predominio de linfocitos, glucosa normal, amilasa normal, hipotrigliceridemia, hipoproteinemia y lactato deshidrogenasa (LDH) baja. Se calcula el gradiente albúmina plasma - líquido ascítico (GASA) > 1,1 g/dL. En el estudio bacteriológico no se obtuvo crecimiento bacteriano. La codificación del estudio BAAR fue 0 y la citología del líquido ascítico fue negativa de células neoplásicas.
En los estudios de las imágenes se reportó en el ultrasonido abdominal un aumento de volumen a nivel abdominal que se corresponde con numerosos quistes de paredes finas, de contenido claro que confluyen entre sí, no se logró determinar el órgano de origen. Impresiona que gran parte de estos se correspondan con el hígado y otros con ambos riñones. El bazo se describió sin alteraciones y no se logró visualizar el resto de los órganos abdominales (fig. 4).
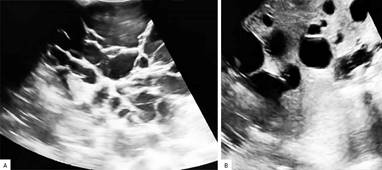
Fig. 4 Ultrasonido abdominal con múltiples quistes de paredes finas y contenido claro en proyección hepática y renal.
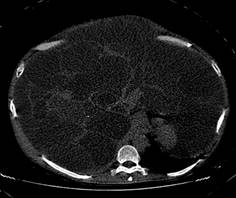
En la tomografía axial computarizada (TAC) simple de abdomen se reportó gran hepatomegalia multiquística que ocupa prácticamente todo el abdomen con riñones poliquísticos (fig. 5).
Con los estudios realizados de la clínica del paciente, sumado a los resultados de laboratorio clínico y estudios imagenológicos, se consideró que se encontraba en el curso de una insuficiencia hepática secundaria a una enfermedad poliquística renal-hepática en estadio avanzado. Se inició tratamiento con diuréticos de asa, albúmina humana, inhibidores de los mineralocorticoides y antimicrobianos; éste último por el riesgo de peritonitis bacteriana espontánea. Paulatinamente el paciente comienza con deterioro de su estado clínico con refractariedad de la ascitis al tratamiento, aumento de los edemas en ambos miembros inferiores y oligoanuria. En complementarios evolutivos se evidencia un empeoramiento de la función hepática, así como de la renal con elevación de la creatinina y la urea; sin criterio de urgencia dialítica al ser valorado por la especialidad de nefrología. Evoluciona hacia la encefalopatía hepática con inestabilidad hemodinámica y fallece. Al realizarse necropsia se aprecian riñones poliquísticos (fig. 6).

Se observaron, además, múltiples quistes hepáticos que deforman la arquitectura de la glándula (fig. 7).
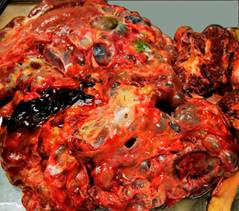
Discusión
La aparición de quistes hepáticos suele cursar de forma asintomática en los estadios iniciales de la enfermedad, pero algunos individuos experimentan manifestaciones crónicas relacionadas con el agrandamiento progresivo de esto, que lleva a la invalidez e impacta severamente en la calidad de vida.10 En un estudio de 239 pacientes con la enfermedad poliquística renal autosómica dominante, Gabow y otros11 encontró que los factores de riesgo asociados a la presencia de quistes hepáticos son la edad avanzada, el sexo femenino y el embarazo, particularmente los embarazos múltiples.
Dentro de las manifestaciones clínicas que se describen en la enfermedad poliquística hepática está el dolor abdominal, el cual suele aparecer por distensión de la cápsula de Glisson. En estadios avanzados de la enfermedad este puede agravarse y aparecer pirosis, saciedad precoz, pérdida de peso y anorexia. Aparece hepatomegalia producto del agrandamiento masivo de los quistes o cuando la glándula hepática se encuentra prácticamente sustituida por los mismos. Dicha hepatomegalia ejerce una compresión sobre el tracto gastrointestinal adyacente con desplazamiento del resto de los órganos intraabdominales, que aumenta así el riesgo de desnutrición por agravamiento de los síntomas.12
Debido al efecto compresivo que ejercen los quistes en estadios avanzados de la enfermedad, ocurren dos procesos que pueden llevar a la aparición de hipertensión portal y a la presencia de ascitis. Estos son: la obstrucción a la salida del flujo sanguíneo en la vena hepática y la obstrucción a la salida del flujo sanguíneo en la vena porta. Este efecto compresivo puede extenderse desde las venas hepáticas hasta la unión con la vena cava inferior, lo cual ocasiona un aumento de la presión en el flujo sanguíneo renal, lo cual favorece el desarrollo de ascitis y edema en miembros inferiores.13
Las opciones de tratamiento para la enfermedad poliquística hepática pueden dividirse en farmacológicas y quirúrgicas. El tratamiento farmacológico se basa en el uso de los análogos de la somatostatina, cuyo efecto principal es la disminución del tamaño de los quistes.14 La aspiración percutánea de los quistes es una opción terapéutica utilizada cuando estos son de gran tamaño, para evitar la aparición de complicaciones. La realización del trasplante combinado hígado-riñón se indica con el objetivo de corregir la desnutrición severa y/o la hipertensión portal así como para mejorar la calidad de vida de pacientes con síntomas severos y en estadios avanzados de la enfermedad.15
En la práctica clínica es poco frecuente la presencia de insuficiencia hepática como causa de muerte en la enfermedad poliquística renal-hepática. Es importante señalar que el desarrollo de la tecnología imagenológica moderna ha facilitado la detección de esta enfermedad y el conocimiento por los médicos. Aunque la presencia de falla hepática se asocia a un pronóstico precario, la detección precoz de la misma puede suponer el inicio de un tratamiento oportuno y apropiado que puede ser beneficioso. Los médicos deben conocer y familiarizarse con esta entidad nosológica ya que constituye un reto en el diagnóstico y terapéutico para la medicina moderna.

