










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
La anemia hemolítica autoinmune (AHAI) se define como el aumento de la destrucción de los eritrocitos producida por autoanticuerpos (auto-Ac) dirigidos contra antígenos de grupos sanguíneos eritrocitarios. La incidencia anual varía de 1-3 en 100 000 a 1 en 25 000 personas. Es una enfermedad altamente heterogénea y afecta a personas de todas las edades, pero es más frecuente en adultos que en niños. Los síntomas y los hallazgos físicos reflejan la destrucción prematura de los eritrocitos con una compensación inadecuada de la médula ósea y el efecto secundario de la hemólisis.1,2,3
Los actores clave en la etiopatogenia de la enfermedad son los auto-Ac con o sin compromiso del sistema de complemento (SC). Sin embargo, se reconocen cada vez más varios efectores inmunitarios celulares, junto con la desregulación de citocinas y la compensación ineficaz de la médula ósea. El diagnóstico suele ser sencillo y se basa en la presencia de anemia hemolítica y evidencia serológica de auto-Ac antieritrocitos detectados por la prueba directa de antiglobulina (PAD).1,2,3
Esta revisión tuvo como objetivo caracterizar las principales variantes serológicas de la anemia hemolítica autoinmune teniendo en cuenta los diferentes tipos de anemias hemolíticas autoinmunes teniendo en cuenta las características fisiopatológicas, manifestaciones clínicas y el diagnóstico de laboratorio.
Métodos
Se realizó una revisión de la literatura en inglés y español, a través de los sitios web PubMed, Science Direct, sí y el motor de búsqueda Google académico de artículos publicados en los últimos 10 años sobre AHAI. Las palabras clave empleadas fueron: anemia hemolítica autoimmune, autoanticuerpos eritrocitarios, autoinmunidad, hemolisis, prueba de antiglobulina directa. El 60,7 % de los trabajos seleccionados fueron artículos originales y de revisión publicados entre los años 2017-2022. Se hizo un análisis y resumen de la bibliografía, se seleccionó los aspectos más importantes referidos al tema.
Análisis y síntesis de la información
Clasificación
La anemia hemolítica autoinmune se ha dividido clásicamente en tipos calientes y frías de acuerdo a la termodinámica de la interacción anticuerpo-antígeno. Históricamente, el término "AHAI caliente" (AHAIC) se ha utilizado para describir la hemólisis mediada por IgG con una interacción antígeno-anticuerpo óptima a la temperatura corporal central, mientras que "AHAI fría" generalmente se refiere a la hemólisis mediada por IgM con una interacción antígeno-anticuerpo óptima a la temperatura periferica.4,5
Con una mejor comprensión de la fisiopatología, el trastorno se puede dividir en cuatro formas serológicas distintas principales: AHAIC, síndrome de aglutininas frías (SAF), hemoglobinuria paroxística “a frigoriˮ (HPF) y la AHAI de tipo mixto (AHAIM) (Tabla 1).4,5,6,7,8
Tabla 1 - Variantes serológicas de la anemia hemolítica autoinmune (AHAI)
| AHAI caliente | IgG | IgG IgG + C3 | IgG | - |
| Síndrome de aglutininas frías | IgM | C3 | No reactivo | > 1: 512 |
| Hemoglobinuria paroxística |
IgG bifásica | C3 | No reactivo | <1:64 |
| AHAI de tipo mixto | IgG + IgM | IgG + C3 | IgG | >1:64 y <1: 512 |
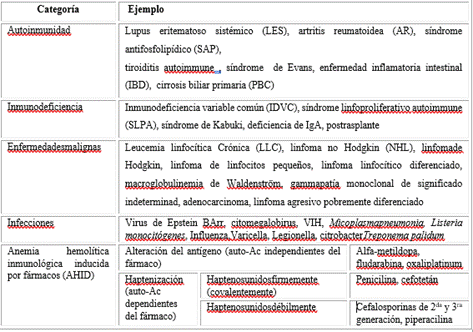
Desde el punto de vista etiológico, también se pueden clasificar en primarias (idiopáticas) y secundarias, siendo esta última un componente o una característica de presentación de un trastorno subyacente al que se atribuye la desregulación inmunitaria. Estos trastornos suelen pertenecer a las categorías de autoinmunidad, inmunodeficiencia, malignidad, infección o exposición a fármacos (Tabla 2).4
El ensayo confirmatorio para el diagnóstico y diferenciación de AHAI es la PAD. La detección inicial se realiza mediante la adición del reactivo antiglobulínico humano o suero de Coombs, un reactivo poliespecífico que contiene anticuerpos antiglobulinas humanas que detecta “in vivoˮ eritrocitos sensibilizados tanto con IgG como con el fragmento C3d del complemento. Si ocurre aglutinación después de la centrifugación de la muestra, esto sería consistente con el entrecruzamiento de eritrocitos y la prueba se considera positiva.
Además, la positividad de PAD se puede clasificar en función de la potencia de la aglutinación y suele oscilar entre 0 y 4+. Si la prueba de detección es positiva, se utilizan antiglobulinas más específicas para diferenciar entre los eritrocitos recubiertos de IgG y C3d de manera similar. También se realiza una caracterización adicional del tipo de anticuerpo mediante la realización de una prueba de antiglobulina indirecta (PAI) o prueba de Coombs indirecta, en la que el suero del paciente o el eluato de los eritrocitos se incuba con un panel de eritrocitos de donantes con características antigénicas conocidas, para detectar la presencia de auto-Ac libres en el suero, plasma o del eluato del paciente.9
AHAIC
La AHAIC es la forma más común de hemólisis inmune y representa el 65 - 70 % de todos los casos de AHAI. Si bien la AHAIC es la forma dominante en los niños, su contribución a la etiología de la anemia hemolítica inmune es menor que en los adultos.10) Se caracteriza por la producción de altas concentraciones de auto-Ac IgG policlonales, generalmente dirigidos contra un componente del complejo proteico de banda 3, fundamentalmente contra antígenos del sistema Rh en menos del 50 % de los casos. La interacción anticuerpo-antígeno óptima ocurre a 37 °C (temperatura corporal central).1,4
Fisiopatología
La hemólisis inmune es principalmente extravascular sin evidencia de activación del SC (solo IgG en el 70 % de los casos) debido a la posición en la membrana eritrocitaria de los antígenos dianas, los cuales están muy separados y no pueden moverse lateralmente debido a su unión al citoesqueleto; por lo tanto, las porciones Fc de los auto-Ac IgG unidos no pueden acercarse lo suficiente entre sí para activar el complemento ya que se necesitan dos moléculas de IgG lo suficientemente cerca para activar C1q e iniciar la cascada de la vía clásica. Además, las moléculas de IgG unidas a dos eritrocitos vecinos no se pueden aproximar debido al potencial zeta que repelen los eritrocitos y los mantienen separados.9
En las ocasiones en que se activa el SC (IgG + C3 en menos del 30 %), esta activación es incompleta producto de la acción de las proteínas y factores inhibidores del sistema, con pobre formación del complejo de ataque de membrana (CAM) C5b-9 que da como resultado hemólisis intravascular. Si el complemento está activado, los eritrocitos se sensibilizan con C3b, los cuales son reconocidos por sus receptores en los macrófagos tisulares y luego se destruyen a través de la fagocitosis mediada por receptores.9,10,11
Los eritrocitos sensibilizados con IgG que viajan a través del bazo, el hígado y la médula ósea son reconocidos por los receptores Fc en los macrófagos tisulares con IgG y actúa como una opsonina y facilita la fagocitosis mediada por receptores. Generalmente son engullidos y digeridos parcialmente por las enzimas lisosomales, lo que deja un eritrocito con una relación de área de superficie/volumen disminuida, que adopta la forma de microesferocitos que es menos deformable y se atrapa fácilmente, luego se destruye en los órganos reticuloendoteliales, especialmente en los senos esplénicos. Con menos frecuencia, se observa eritrofagocitosis completa.12,13
El cuadro serológico típico en AHAIC muestra una PAD positiva con IgG o IgG + C3 en títulos bajos y una PAI generalmente negativa. Esta última es positiva solo si están ocupados todos los sitios antigénicos sobre la membrana del eritrocito y las moléculas de IgG no adheridas se encuentran libres en el suero o plasma del paciente y también en el caso que se encuentre un alo-Ac concomitando junto al auto-Ac. En el caso en que el auto-Ac se encuentre libre, este es polirreactivo y reacciona con todas los eritrocitos del panel celular.4,9
Lo anterior describe la serología de la mayoría de los pacientes con AHAIC, pero alrededor del 3-10 % de los pacientes con un cuadro clínico y de laboratorio compatible con AHAIC tienen una PAD negativa. Esta puede ocurrir por varias razones como:
Número inferior de moléculas de IgG o C3b inferior al rango de detección del suero de Coombs (entre 200-500 moléculas para la IgG o entre 400-1100 moléculas de C3b);
Baja afinidad del auto-Ac opsonizante por el antígeno y se arrastre en el proceso de lavado;
Auto-Ac de clase IgM o IgA por lo que el suero de Coombs comercial no presenta Ac contra estas inmunoglobulinas;
Alteraciones estructurales en la molécula de IgG opsonizante lo que impide la detección por el suero de Coombs;
Inhibición alostérica del suero de Coombs por un sobre exceso de moléculas de IgG sobre la membrana del eritrocito.
Una PAD en microcolumna, el empleo de solución salina de baja fuerza iónica o una citometría eritrocitaria pueden ser útiles para confirmar el diagnóstico si la sospecha clínica es alta.9,14,15,16
El diagnostico confirmatorio de AHAIC se realiza enfrentando el eluato de los eritrocitos del paciente a un panel celular, observándose aglutinación con todas la células.9,17) Es importante resaltar que el 0,1 % de los donantes voluntarios de sangre y hasta el 8 % de todos los pacientes hospitalizados pueden presentar una PAD positiva sin evidencia clínica ni de laboratorio de hemolisis inmune. También se detecta una PAD positiva en pacientes luego de ser transfundidos o posterior a la administración de inmunoglobulina intravenosa con o sin hemolisis significante (Fig. 1).4,18,19
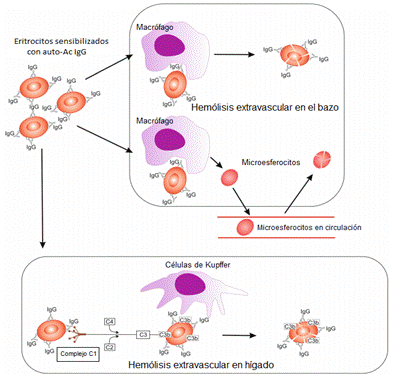 Fuente: Tomado y modificado de Michalak SS, et al. Autoimmune hemolytic anemia: current knowledge and perspectives. Immunity and Ageing. 2020;17:38. DOI: https://10.1186/s12979-020-00208-7
19
Fuente: Tomado y modificado de Michalak SS, et al. Autoimmune hemolytic anemia: current knowledge and perspectives. Immunity and Ageing. 2020;17:38. DOI: https://10.1186/s12979-020-00208-7
19
Fig. 1 - Hemólisis en el curso de la anemia hemolítica autoinmune caliente (AHAIC), mecanismos y sitios de hemólisis. Los eritrocitos sensibilizados con auto-Ac IgG son ingeridos y degradados por macrófagos o destruidos por el mecanismo de citotoxicidad dependiente de anticuerpos en el bazo. Algunos eritrocitos son fagocitados parcialmente y adoptan forma de microesferocitos y regresan a la circulación con la membrana celular más rígida y menos flexible por lo cual son secuestrados en el bazo. La activación débil e incompleta del sistema de complemento por anticuerpos IgG conduce a hemólisis extravascular en hígado.19
Las enfermedades autoinmunes, especialmente el LES, se asocian comúnmente con AHAIC. Hasta el 10 % de los pacientes con LES tienen una AHAIC clínicamente significativa con anemia de moderada a grave, reticulocitosis, hiperbilirrubinemia y lactato deshidrogenasa (LDH) elevada.20) La presencia de anemia hemolítica manifiesta generalmente se asocia con un subgrupo de pacientes con LES que se caracteriza por una edad más temprana de aparición de la enfermedad y una enfermedad más grave con una mayor probabilidad de afectación renal, convulsiones, serositis y otras citopenias.21
Las condiciones con desregulación inmune como la infección por VIH, el SLPA y la IDVC, también se asocian con AHAIC. Aunque la hemólisis inmune relacionada con el VIH generalmente se atribuye a infecciones concurrentes por el virus de Epstein-Barr o el citomegalovirus que generalmente se asocian con aglutininas frías, ocasionalmente se detectan aglutininas calientes. Se observa PAD positiva sin hemólisis hasta en el 18 % de los pacientes con VIH.22,23) Por otra parte, se informa que hasta el 11 % de los pacientes con IDVC tienen alguna forma de citopenia autoinmune, en los cuales el 50 % se presenta antes o en el momento del diagnóstico.24) Además, el 50-70 % de los pacientes con SLPA tienen citopenias autoinmunes, generalmente una combinación de AHAI y trombocitopenia inmune (ITP) que se conoce como síndrome de Evans.25,26
Entre las enfermedades malignas, la LLC es una de las más asociadas con AHAIC en adultos, afecta entre 4-10 % de los casos. Esto generalmente se agrava por el riesgo agregado de atribuido a la quimioterapia, especialmente los análogos de purina como la ludarabina. Alrededor del 90 % de los casos de AHAI en la CLL se deben a auto-Ac IgG policlonales de alta afinidad producidos por clones de células B no malignas dirigidos contra antígenos eritrocitarios.27) Otros mecanismos incluyen la inhibición directa de la eritropoyesis mediada por citocinas por parte de las células T citotóxicas y las células asesinas naturales (NK) en un microambiente inmunitario desregulado inducido por factores solubles secretados por el clon LLC maligno. Además, la positividad de la PAD subclínica se observa hasta el 35 % de los pacientes.28,29
Los fármacos también pueden ser causa de AHAI, destacándose entre ellos los antimicrobianos, antinflamatorios y antineoplásicos. La AHAI inducida por fármacos (AHAID) puede ocurrir con o sin desregulación inmunitaria subyacente, destacándose mecanismos dependientes e independientes del fármaco como lo son la alteración del antígeno y la haptenización de los mismos sobre la membrana eritrocitaria.30
Alteración del antígeno
El fármaco interactúa con la proteína diana en la membrana eritrocitaria y cambia su estructura, por lo que los linfocitos circulantes la considerarían extraña y generarían anticuerpos contra ella.30 A su vez, interactuarían con las copias normalmente expresadas con la subsiguiente destrucción de los eritrocitos. Aquí el fármaco no forma parte del epítopo del antígeno, ni es necesario para la producción posterior de anticuerpos (auto-Ac independientes del fármaco), por lo que su interrupción no garantiza la resolución de la AHAI en estos pacientes.31,32
El ejemplo clásico es la alfa-metildopa, que induce la formación de auto-Ac en aproximadamente el 15 % de los pacientes, mientras que 0,5 a 1 % desarrollan AHID clínicamente significativa. La alfa-metildopa interactúa y altera componentes de la membrana eritrocitaria y los vuelve antigénicos. La respuesta típica de auto-Ac está mediada por IgG; por lo tanto, la mayor parte de la hemólisis es extravascular. No se observa activación del SC ya que los complejos de proteínas diana están muy separados en la superficie de los eritrocitos e impide que las moléculas de IgG unidas activen el complemento. El cuadro serológico típico es una PAD positiva para IgG, negativa para C3b y PAI panaglutinante positivo para IgG.9,31,32,33
Haptenización
Un hapteno es una molécula pequeña que no es antigénica en sí misma, pero cuando se une a una proteína transportadora, aumenta la inmunogenicidad de esa proteína, de modo que los linfocitos circulantes la considerarían extraña. Aquí el epítopo antigénico es un componente esencial del hapteno, a diferencia del ejemplo anterior de alfa-metildopa.9,31
Este mecanismo se subdivide en dos categorías:
Unión covalente del hapteno: el ejemplo clásico es la penicilina, especialmente cuando se administra en altas dosis. El fármaco se adhiere firmemente a un componente de la membrana eritrocitaria y lo hace antigénico. Los auto-Ac IgG específicos se dirigen al complejo de la membrana haptenizada y etiquetan los eritrocitos para la hemólisis extravascular. Nuevamente, el complemento no se activa en este caso. Dada la inclusión del fármaco culpable en el epítopo, los auto-Ac solo reaccionan a las proteínas haptenizadas; por lo tanto, la interrupción del fármaco permite eliminar la hemólisis en pocos días. El cuadro serológico típico es una PAD IgG positivo, C3b negativo, mientras que la PAI estándar solo es positivo si los eritrocitos del panel se tratan previamente con altas concentraciones del fármaco.31,32
Unión débil del hapteno: la evidencia sugiere que el fármaco se asocia vagamente con los antígenos de superficie de los eritrocitos, forma un epítopo que contiene tanto el fármaco como la proteína de los eritrocitos, con auto-Ac que reaccionan tanto a la proteína haptenizada como al fármaco por separado. Las reacciones suelen estar mediadas por IgM, con activación del complemento y hemólisis intravascular. El cuadro serológico típico sería un PAD positiva para C3b (rara vez positivo para IgG) y una PAI negativa que solo se vuelve positivo si se agregan a la reacción tanto el fármaco como una fuente de complemento.31
Características clínicas
Las manifestaciones clínicas asociadas con AHAIC, generalmente, son inespecíficas y se correlacionan con la gravedad de la anemia.
Los pacientes con PAD positiva subclínica son esencialmente asintomáticos, mientras que la mayoría de los pacientes con un diagnóstico claro de AHAIC tienden a tener anemia de moderada a grave que se presenta con fatiga, intolerancia al ejercicio, palpitaciones, disnea de esfuerzo, mareos posturales y dolores de cabeza. Los signos incluyen palidez, ictericia, presión de pulso amplia, taquicardia, precordiohiperdinámico y esplenomegalia de leve a moderada. Los signos de insuficiencia cardíaca de alto gasto, como taquicardia en reposo, crepitantes pulmonares bibasales, edema periférico o distensión venosa yugular, pueden observarse en pacientes con anemia grave, especialmente aquellos con cardiopatía congénita o adquirida subyacente. Además, muchos pacientes con AHAIC secundaria presentaran síntomas y signos relacionados con el trastorno subyacente; por lo tanto, es crucial la búsqueda de posibles marcadores de autoinmunidad, inmunodeficiencia, malignidad o infección subyacentes.4,34
Los hallazgos de laboratorio incluyen:35
Anemia: hemoglobina con un promedio de 70-100 g/L.
Macrocitosis: aumento del volumen corpuscular medio (MCV), secundario a reticulocitosis.
Esferocitosis y aumento de la concentración de hemoglobina corpuscular media (MCHC) debido a la fagocitosis parcial de eritrocitos que reduce la relación entre el área superficial/volumen.
Reticulocitosis: se observa en el 70-80 % de los pacientes, su mayoría superior al 5 % y un índice de producción de reticulocitos >2-3 %. Entre el 20-30 % de los pacientes presentan recuentos normales o levemente reducidos producto de mielosupresión debida a fármacos o infección viral concurrente o anticuerpos que reaccionan con precursores eritrocitarios y reticulocitos; que causan destrucción temprana antes de su liberación.36
LDH elevada y haptoglobina (Hp) disminuida. El 7 % de los pacientes pueden presentar Hp normal o alta, especialmente en quienes la AHAIC es secundaria a autoinmunidad, infección o malignidad subyacentes, elos niveles iniciales de Hp pueden estar elevados, como reactividad de fase aguda. Una combinación de LDH alta y Hp baja tiene una especificidad del 90 % para diagnosticar hemólisis.37,38
Hiperbilirrubinemia indirecta.
Las manifestaciones de hemólisis intravascular mediadas por IgM y por citotoxicidad dependientes de anticuerpos, que incluyen hemoglobinemia, hemoglobinuria, hemosiderinuria o hipocomplementemia; son raras ya que la gran mayoría de la destrucción de eritrocitos es extravascular.1,39
Trombocitopenia o neutropenia en pacientes con infección aguda o AHAI como parte del síndrome de Evans.26
Marcadores inflamatorios elevados como la velocidad de sedimentación globular y la proteína C reactiva, especialmente con autoinmunidad subyacente, infección o malignidad.35
SAF
El SAF o enfermedad por aglutininas frías (AF) representa el 20-25 % de los casos de AHAI. Es causado por la producción de auto-Ac IgM, dirigido a una variedad de antígenos de naturaleza glúcida sobre los eritrocitos.
Las AF existen en un estado de homopentámero que les permite unirse a múltiples eritrocitos al mismo tiempo, evade el potencial zeta y maximiza su capacidad para aglutinar eritrocitos.5,9 A diferencia de las porciones Fc de los auto-Ac IgG, los fragmentos Fc de IgM están unidos entre sí por el péptido “J” y los fagocitos no expresan receptores para ellos; por lo tanto, la opsonización-fagocitosis no contribuye a la hemólisis. Por otro lado, los pentámeros IgM son potentes activadores del complemento, capaces de inducir formas graves, y en ocasiones, potencialmente mortales de hemólisis intravascular.
La gravedad de la hemólisis depende de la concentración de auto-Ac, la temperatura más alta a la que reacciona con el antígeno, el grado de inhibición por los componentes reguladores del complemento en la superficie de los eritrocitos y la especificidad del auto-Ac. La mayoría de las AF son óptimamente reactivas a 0-5 °C, pero las que permanecen reactivas a temperaturas más altas son más patógenas, ya que es más probable que estén activas a la temperatura de las extremidades, especialmente en un ambiente frío.40
Fisiopatología
Las AF se adhieren de manera laxa a los polisacáridos de la superficie de los eritrocitos cuando se alcanza el rango de temperatura requerido, pero en muchos casos la duración del tiempo que la sangre circulante pasa en ese rango de temperatura particular es muy breve, por lo que el anticuerpo se separa sin activación del complemento. A veces, esta unión de corta duración da como resultado la aglutinación de algunos eritrocitos (sin hemólisis) en las puntas de los dedos que causan acrocianosis.
En los casos en que la amplitud térmica de la AF es lo suficientemente alta, todavía es capaz de unirse a los polisacáridos de la superficie eritrocitaria, pero no necesariamente a temperaturas muy bajas. La duración de esa unión es lo suficientemente larga como para permitir la activación del CAM, lo que provoca hemólisis intravascular.40,41
Generalmente se observa hemólisis intravascular solo durante la presentación aguda o la exacerbación de la enfermedad crónica, mientras que la mayor parte de la hemólisis en la forma crónica estable de la enfermedad resulta de la unión de componentes sublíticos, particularmente C3b y en menor grado C4b, que actúan como opsoninas, mediando la fagocitosis de eritrocitos en el hígado por células de Kupffer (hemólisis extravascular).42 La opsonización-fagocitosis mediada por el complemento da como resultado la absorción completa de eritrocitos; por lo que no se observa microesferocitosis (Fig. 2).19,39
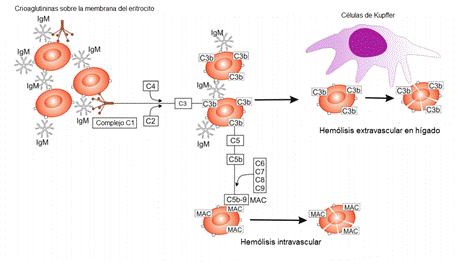
Fig. 2 - Hemolisis en el síndrome de aglutininas frías (SAF). La activación de la cascada del complemento se inicia con el reconocimiento de los antígenos eritrocitarios por las aglutininas frías que causa hemólisis intra y extravascular. La hemólisis extravascular asociada se produce fundamentalmente en hígado por el sistema mononuclear fagocítico. La hemólisis intravascular es el resultado de la formación del complejo de ataque de membrana (MAC). (Tomado y modificado de Michalak SS, et al. Autoimmune hemolytic anemia: current knowledge and perspectives. Immunity and Ageing. 2020;17:38. DOI: https://10.1186/s12979-020-00208-7 19
Las AF se dividen en cuatro tipos principales en función de su especificidad “in vitro” contra polisacáridos de la superficie de los eritrocitos:43,44
Auto-Ac anti-I: reaccionan a los polímeros de aminil-lactosa ramificados presentes en casi todos los eritrocitos adultos y representan la mayoría de las AF patógenas.
Auto-Ac anti-i: reaccionan a los polímeros de aminil-lactosa lineales presentes en los eritrocitos fetales o del cordón.
Auto-Ac anti-J: tienen igual reactividad a los polímeros lineales y ramificados de aminil-lactosa.
Auto-Ac anti-Pr: son poco frecuentes y reaccionan frente a polisacáridos asociados a glucoproteínas tratados con proteasas in vitro.
Características clínicas
El SAF se produce como consecuencia de un proceso infeccioso, paraneoplásico o neoplásico subyacente, pero puede adoptar una forma crónica idiopática conocida como síndrome de crioaglutinina crónico.45
El SAF secundario a una infección se desarrolla, comúnmente, después de una infección por Mycoplasma pneumoniae o EBV. Las AF oligoclonales se forman en 50-75 % de los pacientes dentro de 1-2 semanas de la infección por Mycoplasma pneumoniae, generalmente con especificidad anti-I. La incidencia es mayor en niños y disminuye con la edad.46 No se observa hemólisis clínicamente significativa dados los títulos bajos de auto-Ac y la presencia de fuertes mecanismos inhibidores del complemento, pero una minoría presentará hemólisis grave que requerirá hospitalización y transfusión de hemoderivados. Alternativamente, hasta el 60 % de los pacientes con mononucleosis infecciosa debida a EBV desarrollan anticuerpos anti-i, pero solo el 1 % manifiesta AHAI.9
Otras infecciones asociadas con el SAF incluyen CMV, legionella, citrobacter, influenza y varicela.9,40 La enfermedad se presenta 1-2 semanas después del inicio de la infección primaria y se resuelve dentro de las 2-4 semanas. Los títulos de anticuerpos disminuyen gradualmente y regresan a niveles normales dentro de los 3-4 meses siguientes. A diferencia de la forma aguda, ocasionalmente mortal, pero en última instancia autolimitada, la enfermedad asociada a malignidad adopta una forma más crónica con concentraciones patológicas estables de auto-Ac monoclonales.47
Los pacientes con neoplasias linfoides bien diferenciadas como la LLC y el linfoma de linfocitos pequeños y las discrasias de células plasmáticas (gammopatías) tienden a producir anti-I, mientras que los pacientes con formas agresivas y poco diferenciadas de linfoma tienden a producir aglutininas frías anti-i.
Aunque hay algunas excepciones a esta generalización, es muy probable que un paciente con crioaglutininas anti-i persistentes y sin evidencia de una infección subyacente tenga un linfoma pobremente diferenciado.5 Se cree que la producción de AF relacionada con la malignidad se debe a la estimulación de un clon linfocítico neoplásico, por lo que los títulos tienden a corresponder con la actividad de la enfermedad y pueden usarse como marcadores tumorales o indicadores de recaída.48
Los pacientes con síndrome de AF crónico tienen una gammapatía IgM monoclonal benigna, que suele diagnosticarse en personas mayores con anticuerpos anti-I persistentes. La enfermedad es estable durante muchos años, pero hasta en un 10 % el presunto clon linfoide benigno responsable de la producción de crioaglutininas se transforma en una gammapatía maligna que se presenta con fiebre, leucocitosis, hipergammaglobulinemia, aumento del título de AF, linfadenopatía o hepatoesplenomegalia.5
Los pacientes presentan síntomas generales inespecíficos de anemia y síntomas específicos relacionados con la aglutinación de eritrocitos y la hemólisis intravascular, que incluyen:5,19
Acrocianosis resultante de la aglutinación de eritrocitos en las áreas que se exponen a un ambiente frío. La decoloración púrpura/gris y el dolor asociado se resuelven con el recalentamiento, con hiperemia reactiva mínima. Generalmente, la interacción anticuerpo-antígeno es lo suficientemente larga para permitir la aglutinación, pero no la activación del complemento. Otras manifestaciones cutáneas incluyen fenómenos de Raynaud, livedoreticularis, urticaria o necrosis/ulceración temprana de la piel.
Orina oscura resultante de hemoglobinuria debida a hemólisis intravascular seguida de filtración glomerular de hemoglobina y eventualmente formación de hemosiderina.
Agravamiento de las manifestaciones de la enfermedad por exposición al frío (incluido el consumo de agua fría), estrés, trauma, cirugía y fiebre.
Manifestaciones clínicas de trastornos asociados como linfadenopatía y hepatoesplenomegalia con malignidad, o fiebre y síntomas respiratorios con infección, entre otros.
Los hallazgos de laboratorio incluyen:5,9,19,35
Anemia: los pacientes con producción crónica de crioaglutininas tienen un grado moderado de anemia que se atribuye a la hemólisis extravascular por opsonización C3b, dado que los títulos más bajos de crioaglutininas circulantes no son suficientes para producir una franca hemólisis intravascular en presencia de niveles normales de inhibidores del complemento presentes en la membrana de los eritrocitos. Por otro lado, los pacientes con aglutininas frías relacionadas con infecciones y aquellos con exacerbaciones de enfermedades crónicas debido a la exposición al frío u otros factores estresantes, pueden tener formas graves de anemia atribuidas a hemólisis intravascular franca en presencia de títulos muy altos de AF.
Recuento e índices eritrocitarios inexactos debido a la aglutinación: las muestras de sangre deben calentarse, entregarse en mano al laboratorio y procesarse de inmediato para evitar la aglutinación.
Reticulocitosis: algunos pacientes pueden tener anticuerpos dirigidos a los reticulocitos, o mielosupresión viral o inducida por fármacos, lo que resulta en reticulocitopenia.
Formación de grumos de eritrocitos y rouleaux en el frotis de sangre periférica. No se observan microesferocitosis.
La LDH elevada, la haptoglobina baja; la hemoglobinemia, la hemoglobinuria, la hemosiderinuria y la hiperbilirrubinemia se observan de forma variable, especialmente en la enfermedad crónica aguda o exacerbada.38
PAD C3d positivo e IgG negativo
PAI positiva para IgM monoclonal u oligoclonal reactiva a antígenos polisacáridos (I, i, Pr y otros) a temperaturas entre 0-5°C.
Se observan títulos altos de AF en la mayoría de los pacientes con niveles que van desde 1/2048 hasta 50 000.
Hipocomplementemia (C3 y C4 disminuidos) se observa en muchos pacientes con enfermedad de crioaglutinina debido al consumo constante en la hemólisis intravascular.
La hipergammaglobulinemia se observa en pacientes con enfermedad por crioaglutinina relacionada con la gammapatía monoclonal.49,50
HPF
La HPF se describió por primera vez en asociación con sífilis secundaria o terciaria. Es una rara anemia hemolítica que representa el 1-3 % de los casos de AHAI. Es causa común de AHAI infantil y representa hasta el 30-40 % de los casos en algunos estudios. En adultos es poco frecuente.51 Actualmente se informa como una complicación posinfecciosa o, más raramente, en el contexto de autoinmunidad o malignidad.52
Fisiopatología
La HPF se caracteriza por la producción de un auto-Ac IgG policlonal reactivo en frío, conocido como anticuerpo de Donath-Landsteiner, dirigido contra el antígeno P. El anticuerpo se adhiere a la superficie de los eritrocitos tras la exposición al frío, generalmente en las extremidades, seguido de la activación de C1q, luego la fijación de C2 y C4, pero el proceso generalmente se detiene hasta que la sangre alcanza nuevamente 37°C, el anticuerpo se separa y el resto de la cascada de la vía clásica del complemento se completa, lo que resulta en la formación de CAM y hemólisis intravascular.1,9,52
Características clínicas
La mayoría de los pacientes son niños que desarrollan síntomas dentro de 1-2 semanas del inicio de una infección viral, comúnmente con varicela, pero también se informa con sarampión, parotiditis, EBV, CMV, adenovirus e influenza A. La enfermedad es típicamente aguda, con anemia moderada a grave, ictericia, hemoglobinuria y raramente fenómenos de Raynaud y urticaria. Los síntomas generalmente comienzan a los pocos minutos de la exposición al frío y comienzan a mejorar poco después del recalentamiento. Los anticuerpos suelen persistir hasta 12 semanas, pero la hemólisis suele desaparecer en unas pocas semanas o un mes. Los adultos generalmente tienen un trastorno autoinmune subyacente o una neoplasia linfoide maligna y muestran una forma más crónica de la enfermedad.1,6,53,54
Los hallazgos de laboratorio incluyen:1,6,9,53
Anemia: de moderada a grave en la HPF aguda posinfecciosa que se observa en niños, mientras que es de leve a moderada con exacerbaciones intermitentes en adultos con formas más crónicas de la enfermedad.
Reticulocitosis y rara vez microesferocitosis.
LDH elevada, Hp baja, hemoglobinemia, hemoglobinuria, hemosiderinuria e hiperbilirrubinemia.
Hipocomplementemia.
PAD C3d positivo e IgG negativo (la PAD puede ser negativo según el procesamiento y las variaciones de temperatura).
AHAIM
En estos casos se identifica un cuadro serológico mixto con aglutininas calientes y frías en alrededor del 8 % de los pacientes. En muchos de ellos, el impulsor de la hemólisis es el componente caliente, ya que las AF suelen tener una amplitud térmica baja, siendo reactivas solo a temperaturas bajas (0-20°C) y en muchos casos con títulos bajos. Sin embargo, hay informes de casos de AHAIM verdadera con auto-Ac calientes y aglutininas frías con títulos patológicos de anticuerpos y amplitud térmica alta. La mayoría de estos pacientes tienen un trastorno autoinmune subyacente.1,7,55
La AHAI es una enfermedad muy heterogénea debido a los diversos mecanismos involucrados en su patogenia, causando una enfermedad clínicamente compleja. Además, el tipo y el alcance de la desregulación inmunitaria pueden ser diferentes en cada paciente y cambiar con el tiempo, lo que a menudo determina un curso clínico impredecible.
El diagnostico suele ser fácil, pero los casos difíciles pueden ser un desafío, especialmente si las pruebas inmunohematológicas son negativas o están asociadas con una enfermedad primaria. La definición de cada tipo es fundamental ya que la terapia es diferente y se enfoca más con la comprensión de los mecanismos patogénicos.

