




![Análisis de los compuestos volátiles de la guayaba ácida (Psidium acidum [DC.] Landrum) mediante microextracción en fase sólida del espacio de cabeza](/img/es/next.gif)





 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroduccion
Los gangliósidos son carbohidratos que pertenecen al grupo de los glicoesfingolípidos, que participan en una gran variedad de eventos biológicos que ocurren en las células epiteliales de los vertebrados (Pilco et al, 2019; Blanco et al, 2015; Shimizu et al, 2020). Se ha demostrado que, durante la transformación maligna de la célula, la composición de los gangliósidos cambia cuantitativa y cualitativamente (Pilco et al, 2019; Blanco et al, 2015; González et al, 2016). Por esta razón se encuentran expresados en mayor concentración en los tejidos cancerosos, en comparación con el resto, lo que posibilita el uso de anticuerpos contra él (Gentilini et al, 2016; González et al, 2018; Zheng et al, 2019).
Dado las potencialidades como inmunosupresor de los gangliósidos N-Acetil GM3 (NAcGM3) y N-glicolil GM3 (NGcGM3) y su importancia en el desarrollo tumoral, se han convertido en probados blancos terapéuticos (Zheng et al, 2020; Dorvignit et al, 2019; Uskent et al, 2020; Bjerregaard et al, 2020; Mulens et al, 2009). El gangliósido que contiene la variante N-glicolilada del ácido siálico (NGcGM3) se encuentra sobreexpresado en las células tumorales del cáncer de mama y melanoma (Bjerregaard et al, 2020), convirtiéndose este en uno de los componentes fundamentales de un candidato vacunal contra estas enfermedades, el cual ha mostrado no solo ser eficaz en modelos animales, sino también ha reportado beneficios clínicos palpables en varios ensayos clínicos conducidos (Mulens et al, 2009).
Los gangliósidos NGcGM3y NAcGM3 pueden obtenerse por extracción y purificación de fuentes naturales (eritrocitos de caballo y de perro). La producción de estas moléculas por esta vía se ve limitada por su fuente de procedencia y porque, además, existen regulaciones internacionales muy estrictas para la comercialización de productos provenientes de este tipo de fuente, pues se corre el riesgo de arrastrar contaminantes biológicos, por lo que la obtención de estos gangliósidos mediante la síntesis química resulta favorable, ya que se obtiene como compuesto puro y con una estructura definida (Espinosa et al, 2013). Un intermedio importante en la obtención de esta molécula es el aceptor de lactosa. Para la síntesis de glicoconjugados y oligosacáridos que contengan una unidad de lactosa en su estructura, con frecuencia es necesario proteger las posiciones 3, 4 de la unidad de galactosa que forma la lactosa (Zheng et al, 2019).
En la literatura se encuentra reportada la reacción de protección de estas posiciones con un grupo isopropilidén, de la cual se obtiene el 4-O-(3,4-O-isopropiliden-β-D-galactopiranosil)-α,β-D-glucopiranosido (3´,4´-isopropilidén de lactosa). Como resultado de esta reacción el crudo que se obtiene de la misma contiene, además del 3´,4´-isopropilidén de lactosa, el isómero 4´,6´-isopropilidén de lactosa, los acetales mixtos y la lactosa que no reaccionó (Duclos, R.I. 2000).
Para la separación de 3´,4´-isopropilidén de lactosa se deben realizar dos purificaciones cromatográficas en columnas (Duclos, R.I. 2000). Estas operaciones consumen gran cantidad de tiempo y de reactivos, los cuales son muy costosos y además los mismos tienen uso limitado a escalas superiores (Turner, C. 2006). Este trabajo se centra en la sustitución de la primera purificación cromatográfica por otra operación donde se mejore la economía del proceso y facilite el desarrollo de la operación a escala superior.
Experimentacion
Reactivos
Lactosa para síntesis, Merck
Dimetilfromamida para síntesis, Merck
Cloroformo para síntesis, Merck
Metanol para síntesis, Merck
Ácido metanosulfonico para síntesis, Merck
Trietilamina para síntesis, Merck
Diclrometano para síntesis, Merck
Acetona para síntesis, Merck
Gel de sílice 60 (0,040-0,063) Merck
El material de referencia de 3´,4´-Oisopropilidén-α, β-lactosa y 4´,6´-O-isopropilidén-α, β-lactosa, fueron obtenidos en el laboratorio de síntesis química del Instituto Finlay de Vacunas y purificados por cromatografía de columna con una pureza mayor del 99 %, determinada por RMN 1H (Espinosa et al, 2013).
Control de la calidad del proceso
En todo el proceso de síntesis se utilizó la cromatografía de capa delgada para el control del mismo. Para esto se emplearon placas pre-elaboradas de gel de sílice 60 GF254 (Merck) de 0,1 mm de espesor, en las cuales fueron aplicadas las muestras obtenidas en cada etapa y el material de referencia. El sistema de disolventes que se utilizó fue acetato de etilo: 2-propanol: agua (3:2:1; V:V:V). Los cromatogramas se visualizaron por tratamiento con una disolución de ácido sulfúrico al 5 % en etanol seguido por calentamiento a 105 ºC durante 5 min.
Síntesis del 3´,4´-isopropilidén de lactosa
La reacción se realizó en un reactor de 1 L de capacidad provisto de camiseta, agitador mecánico, condensador y un termopar. Primeramente, se suspendieron 50 g de lactosa en 500 mL de dimetilformamida mediante agitación. Cuando esta suspensión alcanzó la temperatura de 85 ºC con ayuda de termostato, se añadieron 160 μL de ácido metanosulfónico y 42,3 mL de dimetoxipropano. El curso de la reacción se chequeó por cromatografía de capa delgada con el sistema de disolventes acetato de etilo: 2-propanol: agua en proporción 3:2:1. Transcurridos los 45 minutos de reacción se retiró el calentamiento y se enfrió a temperatura ambiente. Cuando se logró esta condición, se adicionaron 10 mL de trietilamina para neutralizar la reacción. Una vez terminada se eliminó la mezcla de disolventes por roto evaporación, usando un rotoevaporador de 2 L de capacidad con una temperatura de baño de 40 ºC y una presión de 556 mbar (Espinosa et al, 2013).
Extracción líquido-líquido
Al crudo de la reacción de síntesis de 3’,4’-isopropiliden de lactosa se le adicionaron 100 mL de agua y se agitó hasta total dilución.
En un embudo separador de 500 mL con agitación mecánica se colocó la fase acuosa y se le adicionaron 50 mL de diclorometano, se agitó por un tiempo de 5 minutos y luego se separaron ambas fases. Esta operación se realizó cinco veces más para extraer la mayor cantidad de acetales mixtos. La fase acuosa se evaporó a sequedad con ayuda de un rotoevaporador de 2 L de capacidad con una temperatura de baño de 50 ºC y una presión de 72 mbar, hasta obtener un sólido espumoso. La operación se controla por cromatografía de capa delgada para comprobar que el sólido obtenido contiene lactosa y la mezcla de isómeros y en la fase orgánica se encuentra los acetales mixtos extraídos (Espinosa et al, 2013).
Estudio de las variables de operación en la extracción líquido-líquido
Se estudió el efecto de las variables velocidad de agitación y relación entre el disolvente orgánico y la fase acuosa para esta etapa de extracción. Para esto se realizó un diseño de experimento 31•21, con una réplica en cada punto, es decir una repetición del experimento básico, el cual se muestra en la tabla 1. Como niveles inferiores para las variables a estudiar se fijaron 300 rpm de velocidad de agitación y la relación entre el disolvente orgánico y la fase acuosa 0,5:1.
Los resultados fueron analizados con la ayuda del programa estadístico Statgraphics Centurion en su versión XV.II., donde se realizó el análisis de varianza para un 95 % de confianza. Se obtuvo además el modelo ajustado y la superficie de respuesta.
Extracción líquido-sólido
Esta extracción se realizó en el mismo balón donde se evaporó la fase acuosa de la operación anterior. Al sólido espumoso obtenido en la evaporación de la fase acuosa se le adicionaron 75 mL de acetona, le fue conectado un agitador mecánico y se agitó por un tiempo de 15 minutos. Ambas fases se separaron por filtración a vacío en un embudo Büchner con placa y papel de filtro. A la fase sólida obtenida se le realizó siete veces más esta operación con el objetivo de extraer la mayor cantidad posible de la mezcla de isómeros. La operación se controló por cromatografía de capa delgada para comprobar que la fase líquida contiene la mezcla de los isómeros 3´,4´y 4´,6´-isopropilidén de lactosa, la misma se evaporó a vacío con ayuda de un rotoevaporador de 2 L de capacidad con una temperatura de baño de 50 ºC y una presión de 11 mbar (Espinosa et al, 2013)
Estudio de las variables de operación en la extracción líquido-sólido
Para estudiar el efecto de las variables velocidad de agitación y relación líquido-sólido en esta etapa, se realizó un diseño de experimento 31 •21, con una réplica en cada punto, es decir una repetición del experimento básico. Como niveles inferiores para las variables a estudiar se fijaron 300 rpm de velocidad de agitación y la relación líquido-sólido 3:1.
Tabla 2 Efecto de la relación líquido-sólido y la agitación sobre la separación de la lactosa de la mezcla de isómeros.

Los resultados fueron analizados con la ayuda del programa estadístico Statgraphics Centurion en su versión XV.II., donde se realizó el análisis de varianza para un 95 % de confianza. Se obtuvo además el modelo ajustado y la superficie de respuesta.
Determinación del número de extracciones líquido-sólido
Con el objetivo de definir el número de extracciones a realizar en ambas operaciones se realizaron cuatro extracciones más que las establecidas según el procedimiento, para encontrar el punto de agotamiento del producto a extraer. Con los resultados obtenidos se realizó la curva de agotamiento para cada uno de los productos a extraer, utilizando para ello el programa Curve Expert Professional.
Purificación cromatográfica
Se tomó una columna cromatográfica a la cual se le adiciona sílica gel 60 como fase estacionaria y se le comienza a adicionar mezcla de disolventes cloroformo metanol en proporción 50:1, para compactar la misma. Posteriormente, se adicionó el crudo de reacción previamente disuelto en metanol e impregnado en sílica gel 60. Luego se comienza a adicionar la mezcla de disolventes variando la polaridad de la misma, para obtener una primera fracción donde se encuentran los acetales mixtos y posteriormente la fracción de la mezcla de los isómeros. Esta última fracción se evaporó hasta peso constante.
RESULTADOS
Influencia de la relación entre el disolvente orgánico y la fase acuosa y la velocidad de agitación en la extracción líquido-líquido
En la tabla 3 se presentan los resultados alcanzados en el plan experimental descrito anteriormente, donde se aplicó el método gravimétrico para determinación de los mismos. Como variables respuestas se obtuvo la masa extraída de acetales mixtos y la masa de la mezcla de isómeros y lactosa al final de la extracción.

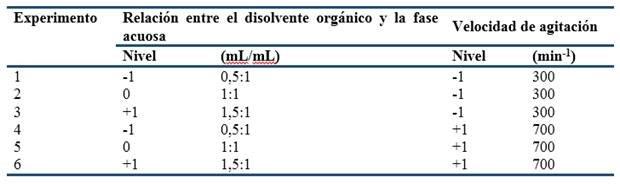
En las Figura 1 y 2 se muestran el diagrama de Pareto y la superficie de respuesta resultantes del análisis estadístico de estos resultados para la masa de acetales mixtos.

De este análisis se encontró como variables significativas (p < 0,05) la relación entre el disolvente orgánico y la fase acuosa y su interacción con la velocidad de agitación, ambas con efecto positivo. Esto demostró que al aumentarse la velocidad de agitación y la relación entre el disolvente orgánico y la fase acuosa se logra extraer una mayor masa de acetales mixtos del crudo de la reacción. Se comprobó a partir de cromatografía de capa delgada que en la fase orgánica solamente había acetales mixtos, no arrastrándose en esta operación el producto de interés.
De este mismo análisis se obtuvo el polinomio ajustado para el efecto de la relación entre el disolvente orgánico y la fase acuosa y de la velocidad de agitación sobre la masa de acetales mixtos en el proceso de extracción (ecuación 1)
Este mismo análisis se realizó para la masa extraída de isómeros y lactosa residual. En la Figura 3 se muestra el diagrama de Pareto correspondiente.

En el diagrama de Pareto se muestra que solamente una variable es significativa (p<0,05): la relación entre el disolvente orgánico y la fase acuosa con efecto negativo. Esto demuestra que al aumentarse la relación entre el disolvente orgánico y la fase acuosa se obtiene menor cantidad de mezcla de isómeros y lactosa residual, debido a que se extrae mayor cantidad de acetales mixtos del crudo obtenido en la reacción, lo que se comprobó a partir de cromatografía de capa delgada como se explicó en el análisis de la influencia de estas variables sobre la masa de acetales mixtos.
De todo esto se tiene como conclusión que, la mejor condición de operación para esta extracción es trabajar con el nivel 1 de la relación entre el disolvente orgánico y la fase acuosa y con el nivel 1 de la velocidad de agitación.
Determinación del número de extracciones a realizar
En la Figura 4 se muestra la curva de agotamiento de los acetales mixtos, la misma se obtuvo de graficar las 10 extracciones realizadas a las mejores condiciones encontradas en esta operación. El modelo ajustado se representa en la ecuación 1, donde n representa el número de extracciones realizadas. El coeficiente de correlación obtenido fue 0,99.

 (2)
(2)
Mediante esta ecuación fue posible predecir el número de extracciones que son necesarios realizar para extraer totalmente los acetales mixtos. También es posible calcular la cantidad máxima de acetales a extraer y el porcentaje de agotamiento que se obtiene al cabo de un determinado número de ellas. De esta manera se determinó que en ocho extracciones se separaba el 94,3 % de los acetales mixtos presentes en el crudo de reacción. De realizar un mayor número de extracciones se incurriría en un mayor gasto de disolvente, lo que afectaría la economía del proceso y además se tuvo en cuenta que el 5,7 % de acetales que no se remueve, no influye en los pasos posteriores del proceso.
Influencia de la relación líquido-sólido y la velocidad de agitación en la extracción líquido-sólido
Los resultados obtenidos para la extracción líquido-sólido se muestran en la tabla siguiente, donde se aplicó el método gravimétrico para determinación de los mismos. La variable respuesta analizada fue la masa de mezcla de isómeros 3´,4´y 4´,6´-isopropilidén de lactosa.

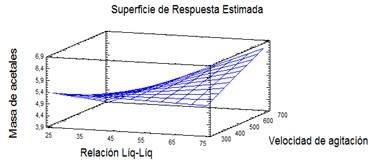
El análisis estadístico de estos resultados brindó el diagrama de Pareto y la superficie de respuesta que se muestran en las Figuras 5 y 6.

Fig. 5 Significación de la relación líquido-sólido y la velocidad de agitación sobre la extracción de la mezcla de isómeros.
De este análisis se encontró que las dos variables estudiadas resultaron significativas, relación líquido-sólido con efecto positivo por lo que se decidió seleccionar el nivel 1 y la velocidad agitación con efecto negativo. La significación negativa de la velocidad de agitación puede explicarse teniendo en cuenta que el trabajo experimental se desarrolló en un balón y al aumentar la velocidad del nivel -1 al 1 aumentó considerablemente el vórtice, este fenómeno creado no es deseado por su efecto negativo sobre la calidad del mezclado. O sea que, se debe trabajar a una velocidad de agitación que garantice una adecuada calidad del mezclado para favorecer la separación de la mezcla de los isómeros de la lactosa residual.
De este mismo análisis se obtuvo el polinomio ajustado que describe el proceso de extracción líquido-sólido (ecuación 3)
Determinación del número de extracciones a realizar
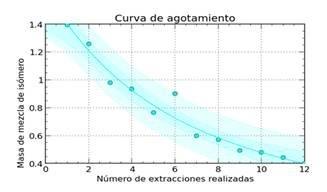
En la Figura 7 se muestra la curva de agotamiento de la mezcla de isómeros 3´,4´ y 4´,6´ isopropilidén de la lactosa, la misma se obtuvo de graficar las 12 extracciones realizadas con las mejores condiciones encontradas en esta operación. La ecuación 4 representa el modelo ajustado donde n es el número de extracciones realizadas. El coeficiente de correlación obtenido fue 0,98.
 (4)
(4)
A partir de la ecuación 4 está claro que no se pueden separar completamente la mezcla de isómeros de la lactosa en un tiempo finito. No obstante, empleando esta ecuación, se estimó la masa de isómeros extraída en las extracciones posteriores al número 12 hasta no apreciar variaciones en esta en el orden de las centésimas, este límite se fijó teniendo en cuenta la precisión del método gravimétrico utilizado para medir la masa de la mezcla de isómeros. Como conclusión de este análisis se determinó que en 12 extracciones se separaba el 65 % de la mezcla de isómeros presentes en el crudo de la reacción. Se determinó además que de realizar un mayor número de extracciones aumentaría el consumo de disolvente y las cantidades que se obtendrían de masa de isómeros serian pequeñas, por lo que se vería afectada la economía del proceso.
Comparación de los dos procedimientos de purificación
Para comprobar la posibilidad de sustituir la primera purificación cromatográfica por una extracción líquido-líquido y una extracción líquido-sólido, se realizó la comparación de dos lotes: uno donde los pasos de purificación coinciden con lo descrito en la literatura, una purificación cromatográfica utilizando como disolventes cloroformo: metanol y como fase estacionaria sílica gel 60 y otro donde se realizaron los pasos propuestos, una extracción líquido-líquido y una extracción líquido-sólido, ambas operaciones utilizando las mejores condiciones encontradas en los epígrafes anteriores. En ambos casos se obtiene la mezcla de los isómeros 3´,4´ y 4´,6´isopropilidén de lactosa.
En la tabla 5 se aprecian las cantidades obtenidas siguiendo las dos variantes de purificación y en la tabla 6 se muestra una comparación de tiempo y volúmenes de disolventes consumidos en ambos métodos.


Como se observa en la tabla 5 la masa de mezcla de isómeros que se obtuvo por la vía propuesta es superior a la obtenida por la vía reportada en la literatura (Duclos, R.I. 2000). Además, esto se logró con una sensible disminución del tiempo de operación y del consumo de reactivos (tabla 6), lo que impacta directamente en la económica del proceso.
Conclusiones
Se encontraron las mejores condiciones en los intervalos estudiados en ambas extracciones que permiten obtener las mayores masas de los productos en cada uno de ellas. En la extracción líquido-líquido las condiciones adecuadas de operación son: relación entre el disolvente orgánico y la fase acuosa 1,5:1, velocidad de agitación 700 rpm y realizar ocho extracciones. En la extracción líquido-sólido las condiciones adecuadas de operación son: relación líquido-sólido 9:1, velocidad de agitación 300 rpm y realizar 12 extracciones.
El método de purificación propuesto de una extracción líquido-líquido y una líquido-sólido permiten obtener cantidades superiores de la mezcla de los isómeros 3´,4´ y 4´,6´isopropilidén de lactosa a las logradas empleando purificaciones cromatográficas, en un tiempo menor y con una notable reducción del consumo de reactivos.

