










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.80 n.2 Ciudad de la Habana abr.-jun. 2008
ORIGINAL
Síndrome de Ehlers Danlos, ¿subregistro clínico en ortopedia pediátrica?
Ehlers Danlos syndrome, a clinical subregister in pediatric orthopedics?
George N. García Rodríguez,I Maricel León Jorge,II Tania Palacios Castillo,III Jorge Luis López CabelloIV
IEspecialista de I Grado en Ortopedia y Traumatología. Instructor. Hospital Pediátrico Docente «José Martí». Sancti Spiritus, Cuba.
IIEspecialista de I Grado en Medicina General Integral. Instructor. Hospital Pediátrico Docente «José Martí». Sancti Spiritus, Cuba.
IIIEspecialista de I Grado en pediatría. Instructor. Hospital Pediátrico Docente «José Martí». Sancti Spiritus, Cuba.
IVEspecialista de I Grado en Ginecología. Instructor. Hospital Pediátrico Docente «José Martí». Sancti Spiritus, Cuba.
RESUMEN
INTRODUCCIÓN. El comportamiento del síndrome de Ehlers Danlos hizo que se interpretara como un subregistro clínico, poco conocido y con escasas referencias. Esta genodermatosis generalmente está condenada a no tener un tratamiento específico. El objetivo del presente estudio fue, principalmente, describir la morbilidad de este síndrome, desde el punto de vista ortopédico.
METÓDOS. Se realizó un estudio descriptivo prospectivo de 5 años, que incluyó a todos los pacientes con síndrome de Ehlers Danlos, atendidos en la consulta de ortopedia y traumatología entre julio de 2001 y julio del 2006, en el Hospital Pediátrico Docente «José Martí» (Sancti Spiritus, Cuba). Se consideró un tiempo mínimo de seguimiento de 6 meses para la validación de los resultados.
RESULTADOS. Fueron estudiados 41 pacientes afectos de 72 enfermedades de origen ortopédico. La frecuencia estuvo próxima a 1,7 enfermedades por paciente, con predominio no significativo del sexo femenino (n = 24). Uno de los antecedentes perinatales más importantes fue la presencia de grados diversos de displasia de la cadera. La presencia de otras afecciones no ortopédicas no fue significativa. Los principales hallazgos ortopédicos fueron el pie plano flexible (37), el genus recurvatum (11) y la cifoescoliosis (9). La cirugía estética y la cirugía correctora ortopédica fueron las más utilizadas.
CONCLUSIONES. La dispensarización de esta enfermedad y su tratamiento oportuno es un método de control eficaz que ayudaría a evitar la degeneración articular, generalmente antesala de la osteoartrosis.
Palabras clave: Síndrome de Ehlers Danlos, hiperlaxitud articular, vitamina C, escala de Beighton, criterios de Brighton, Proyecto Genoma Humano, genodermatosis.
ABSTRACT
INTRODUCTION. The behavior of Ehlers Danlos syndrome caused it to be interpreted as a clinical subregister, little known, and with a few references. This genodermatosis is generally condemned not to have a specific treatment. The objective of this study was mainly to describe the morbidity of this syndrome from the orthopedic point of view.
METHODS. A 5-year prospective and descriptive study was conducted among all the patients with Ehlers Danlos syndrome that were seen at the Orthopedics and Traumatology department of "José Martí" Teaching Pediatric Hospital (Sancti Spiritus, Cuba) from July 2001 to July 2006. A minimum follow-up time of 6 months was considered for the validation of the results.
RESULTS. 41 patients affected with 72 diseases of orthopedic origin were studied. The frequency was approximately 1.7 diseases per patient, with a non significant predominance of females (n = 24). One of the most important perinatal antecedents was the presence of diverse hip dysplasia degrees. The presence of other non-orthopedic affections was not remarkable. The main orthopedic findings were the flexible flat foot (37), the genus recurvatum (11) and kyphoscoliosis (9). Aesthetic surgery and the corrective orthopedic surgery were the most used.
CONCLUSIONS. The categorization of this disease and its timely treatment is an efficient method of control that would help to prevent the articular degeneration that generally precedes osteoarthritis.
Key words: Ehlers Danlos syndrome, articular hyperlaxity, vitamin C, Beighton's scale, Brighton's criteria, Human Genoma Project, genodermatosis.
INTRODUCCIÓN
Se adjudica el nombre de síndrome de Ehlers Danlos (SED) a un grupo heterogéneo de alteraciones genéticas de la estructura y síntesis del colágeno y del tejido conectivo (genodermatosis). Ehlers (1901) y Danlos (1908), fueron los primeros autores en correlacionar la hipermovilidad articular en un síndrome que presentaba una serie de anormalidades de la piel y del tejido conjunto de otros órganos,1 aunque algunos estudios previos se habían hecho, atribuidos a Tschernogobow in 1892,2,3 y a Hipócrates, que había descrito la afección en algunas poblaciones del Mar Caspio. Se recoge en la historia también, casos de músicos virtuosos que padecieron esta enfermedad como Paganini y Rachmaninoff.4
El SED engloba a un grupo heterogéneo de cerca de 11 asociaciones patológicas, todas relacionadas con una alteración genética de la estructura, la síntesis del colágeno y del tejido conectivo,5 unificados a partir del congreso de genética humana de Berlín, aunque a largo plazo, requirió una nueva reunificación de ellos para hacerlos mas funcionales. En 1997, y como resultado del grupo de trabajo liderado por Beighton vio la luz la nueva clasificación, vigente en la actualidad que quedó conformada los por 6 grupos que consta de:6-26
- Clásica (antiguos grupos l y ll).
- Hipermovilidad (antiguo grupo lll).
- Vascular.
- Xifoescoliosis.
- Artrocalasia.
- Dermatosparaxis.
De cualquier forma, en la literatura actual se siguen publicando ambas clasificaciones a la hora de citar este síndrome.
Implica la formación anormal de tejido conectivo, el cual forma la armazón que da soporte y sostén a casi todos los órganos y sistemas que constituyen el cuerpo humano. El colágeno es una proteína altamente distribuida en todo el organismo. Los progresos obtenidos en el proyecto genoma humano y otros avances en la ingeniería genética y molecular, muestran que cerca de 29 genes contribuyen a la formación de su estructura, localizados en 15 de los 24 cromosomas; estos están responsabilizados con la formación de, al menos, 19 formas de la molécula de la proteína colágena.26-30
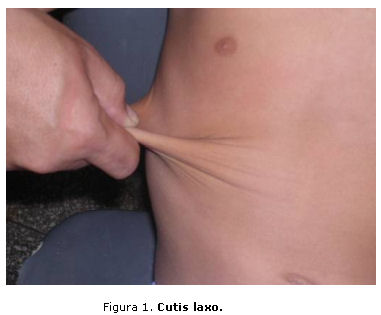
El síndrome se caracteriza por la afectación de la piel, articulaciones y vasos sanguíneos, esto se manifiesta principalmente como hipermovilidad articular, hiperextensibilidad y fragilidad cutáneas. En general la piel es hiperelástica, frágil y se daña con facilidad, lo que tiene como dificultad para la cicatrización; además, se presenta hiperextensibilidad de las articulaciones, con tendencia a las luxaciones frecuentes, fragilidad de los vasos sanguíneos (figuras 1 y 2). Otras complicaciones son recurrentes como la aparición de hernias recidivantes, várices precoces -con paredes frágiles, difíciles de suturar-, hemorroides y varicocele.5-21


El objetivo del presente trabajo estuvo dirigido a la necesidad de determinar la morbilidad que desde el punto de vista ortopédico presenta el SED, que en ocasiones no pasa de ser un subregistro clínico, poco conocido y condenado al no tener un tratamiento específico.
MÉTODOS
Se realizó un estudio prospectivo que abarcó un período de 5 años en todos los pacientes atendidos en la consulta de ortopedia y traumatología entre julio de 2001 y julio del 2006, con diagnóstico de SED, en el hospital pediátrico docente José Martí, de Sancti Spiritus, Cuba.
La validación de los resultados de la investigación se dio con un tiempo mínimo de seguimiento en la consulta externa de 6 meses. El universo de estudio coincidió con la muestra, constituida por 41 pacientes.
Criterios diagnósticos del SED
Para el inicio de esta investigación se dio la tarea de crear un protocolo previo, basado en la evidencia clínica, que incluyó la aplicación del score de Beighton, y los criterios de Brighton en los casos de hipermovilidad, por razones de espacio no se incluyó la totalidad de su contenido, bien definidos en toda la literatura médica.2,8,26
Solo cuando existió la duda razonable para la inclusión o no en el estudio de un caso, se procedió a la realización de biopsia de piel, útil en algunos subtipos mencionados en la introducción de este artículo.
Criterios de inclusión: pacientes que reunieron los requisitos para estar incluidos dentro del síndrome y asistieron a la consulta en el plazo fijado en la investigación.
Criterios de exclusión: pacientes que a pesar de presentar algunas manifestaciones clínicas, no reunieron los requisitos para el diagnóstico de SED. Cuando a pesar de reunir los requisitos para, existió la oposición manifiesta a no ser incluido en el estudio por parte del paciente o tutor, así como el abandono de la investigación.
Al incluir los datos y fotos que se muestran en esta investigación se tuvo en cuenta el consentimiento informado y demás especificaciones normadas para la investigación científica en niños. Por las características de esta institución solo se incluyeron pacientes hasta 18 años.
En la aplicación estadística se utilizaron los programas SPSS/ PC para Windows versión 7.5 y el paquete estadístico de Epinfo versión 6.04 para el cálculo de la muestra.
RESULTADOS
Aun sin la intención de clasificarla en cada subtipo cualquiera que se de a esta tarea, comprenderá lo difícil de esta faena, sobre todo en edades pediátricas donde es poco frecuente la aparición de algunas complicaciones que pueden dar la pauta para la sospecha de este síndrome. Al final de la investigación fueron incluidos 41 pacientes que reunieron los requisitos de participación. Se hizo biopsia de piel en 3 casos, mediante microscopia óptica, lo que mostró como afectación predominante la disminución en la cantidad y desorganización de los haces fibrosos. La fibra colágena anormal es incapaz de organizarse en cordones, lo que produce una matriz colágena defectuosa siendo esta proteína un componente esencial de los tejidos conectivos distensibles.
No se incluyeron en el trabajo como parámetros, los grupos de edades, pues la asistencia a la consulta esta determinada de forma aleatoria recurriendo a la necesidad subjetiva de salud de la familia y el paciente; además, de ser el SED un síndrome genético que se manifiesta en diferentes etapas durante el transcurso de la vida, por el contrario referente al sexo, se observó una ligera desproporción, la incidencia en el femenino fue mayor que en el masculino (24 de 41 pacientes).
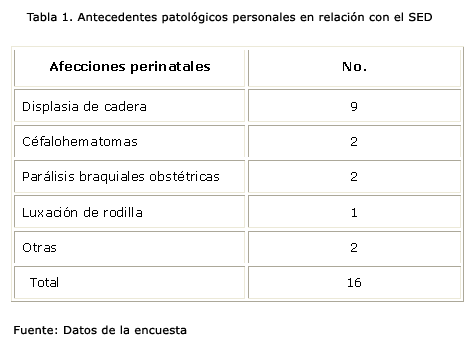
En cuanto a los antecedentes perinatales solo se constató grados variables de displasias de cadera, pero sin llegar a la artroclasia como entidad propiamente descrita. Esta fue el principal antecedente perinatal que se pudo recoger en la muestra en relación al SED, 9 de 16 casos, como muestra la tabla 1. Seguidas de otras de menor cuantía pero que pudieran estar en relación directa o indirecta con el síndrome, como son las lesiones por tracción del plexo braquial y los cefalohematomas.

La indagación de la asistencia de estos pacientes a otras especialidades antes y durante la investigación reveló. Primeramente. Que en esta etapa no han ocurrido las potenciales complicaciones descritas en este síndrome y la incidencia por enfermedades no se diferencia mucho del patrón habitual del resto de la población, lo que quedó como causa primaria, la cirugía estética correctora el principal motivo de recurrencia al quirófano dentro de las afecciones no ortopédicas (tabla 2).

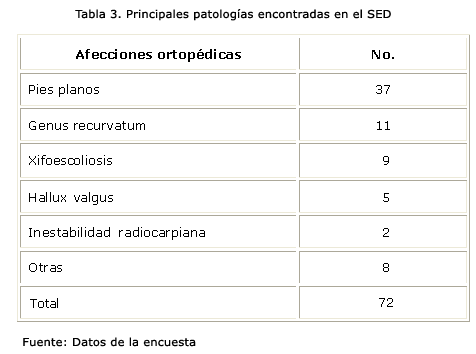
A la hora de atender la morbilidad del sistema osteomioarticular, fueron recogidas 72 enfermedades (tabla 3), donde se aprecia que el pie plano fue la más frecuente en el SED, con 37 de los 41 pacientes en el medio, seguida del Genus recurvatum, apreciada correctamente en los criterios de esta enfermedad.

La tabla 4 se expone, como consecuencia lógica de lo anteriormente expresado, que la cirugía correctora del pie plano es la causa más frecuente recurrida.

DISCUSIÓN
El SED una enfermedad de causa genética, que depende de su tipo, el patrón autosómico dominante, que es lo más frecuente. Otras formas clínicas incluyen patrones de herencia como son autosómico recesivo y recesivo ligado al cromosoma X. La presentación global de todos los tipos de SED son aproximadamente 1 en 5 000 nacimientos mundiales. El predominio de los 6 tipos difiere dramáticamente. Los más común son la forma clásica y la hipermovil, aproximadamente el 90 %. Por suerte otras formas de presentación son muy raras. La dermatosparaxis solo ha sido descrita en una decena de infantes a nivel mundial.2,26
La realización de la biopsia mediante microscopia óptica no mostró resultados mas allá de los esperados mediante el examen clínico, sirvió tal vez, para mostrar una cicatriz hipertrofia en el área de la cirugía derivada de la mala cicatrización.
A pesar de que las distintas investigaciones coinciden en afirmar la igualdad de presentación del SED entre sexos,2,26 la diferencia no significativa a favor del sexo femenino en el trabajo puede estar originada en estas etapas por la desigual en la preferencia de la realización de actividades físicas en individuos jóvenes, que contribuye mediante el control muscular a mantener un tono adecuado. Otra arista podría ser la importancia que las pacientes jóvenes, padres de estas dan al aspecto estético en contraparte al sexo masculino a la hora de acudir a consultas médicas.
Algunos autores,2,26 al igual que los resultados obtenidos, muestran que las displasia de cadera en grados variables pueden considerarse como indicadores tempranos de hiperlaxitud, aunque el resto puede tomarse como causas indirectas, ya que la enfermedad disminuye la capacidad de resistencia al trauma obstétrico.
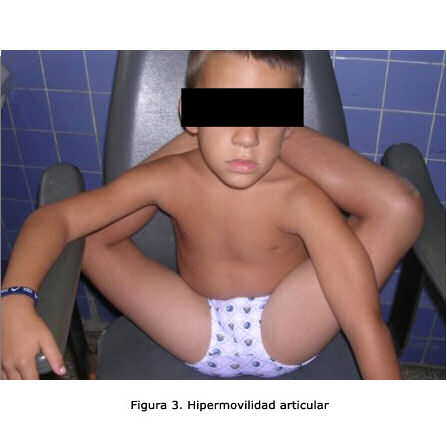
Por lo común la aparición de afecciones no ortopédicas, se retrasa en estas edades. La morbilidad no difiere del resto poblacional. Las características fenotípicas incluyen un rostro distintivo que no pocos autores enfatizan como criterios clínicos de peso en el síndrome (figura 3). La cirugía correctora del pabellón auricular fue la más recurrida y se observó el valor que algunos pacientes le dan en estas edades, al aspecto estético que le ocasiona su enfermedad.8,9
Muchos trabajos han demostrado la disminución de la respuesta propioseptiva de los receptores del SOMA, lo que acarrea como resultado la adopción de posturas biomecánicas articulares inadecuadas y favorece la aparición temprana de cambios degenerativos. Clínicamente puede presentarse desde formas leves a severas, en dependencia del tipo.5-21
Los resultado en cuanto a las entidades ortopédicas (72 afecciones) coinciden con el resto de la literatura,5-11 la frecuencia fue de casi 1,7 por pacientes, localizadas fundamentalmente en el miembro inferior. Estas son las extremidades más afectadas y potencialmente más expuestas a daños, al concentrarse sobre esta las principales consecuencias por la inestabilidad, hiperlaxitud y el consiguiente deterioro articular progresivo, que unido a las alteraciones biomecánicas de la marcha es causa muchas veces de la necesidad de acudir a consulta con el diagnóstico de «pie plano» y «genus recurvatum».
Al concluir el trabajo se pudo encontrar un número mayor de individuos diagnosticados como Ehlers Danlos, que en anteriores etapas al inicio de esta pesquisa hace pensar en un subregistro de esta enfermedad. A pesar de que constituye un síndrome eminentemente clínico, se espera que en los próximos años mejore y facilite el diagnóstico genético de esta afección.
El tratamiento del SED, desgraciadamente arrastra el adagio de no tener un tratamiento propiamente dicho, limitado a tratar sus posibles complicaciones. Se recomienda la dispensarización de estos pacientes; que permitirá brindarle un correcto conocimiento de su entidad y un consejo genético oportuno.
La rehabilitación temprana está dirigida a tratar los grupos musculares específicos, para evitar la aparición de deformidades tempranas; así como el control de peso, la sustracción de actividades físicas extremas y el uso de calzado corrector adecuado u otro tipo de ortesis.
La vitamina C se ha venido utilizando como tratamiento oral, no se recomienda otros "protectores del cartílago" como el sulfato de glucosalina, por encontrar evidencia de efectos colaterales como hemorragias y no contar con suficientes estudios en niños.
La cirugía correctora temprana debe contemplar algunos aspectos como son la elección de técnicas de mínima invasividad, lo que evita problemas a la hora de la evolución de la herida y la aparición de cicatrices antiestéticas mal valoradas por los pacientes y el precepto que una corrección a tiempo ayudaría a evitar una degeneración articular, generalmente la antesala de una futura osteoartrosis.
REFERENCIAS BIBLIOGRÁFICAS
1. Danlos M. Un cas de cutis laxa avec tumeurs par contusion cronique des coudes et des genoux (xanthome juvenile pseudo-diabetique de MM. Hallopeau et Marce deLepinay). Bull Soc Français Dermatol Syphilig. 1908;19:70-72.
2. Steiner R, Bradley G, Pepin M. Ehlers-Danlos Syndrome. 2006. [Citado Mayo 24 de 2007] disponible en: http://www.emedicine.com
3. Tschemogubow AN. Ein fall von cutis laxae. Protokoly Moskowskawo Venereoologitscheskawo i Dermatlogitscheskawo. Obtschestwa. 1891/1892;1:23-29.
4. Ho N, Park S, MaragH KD, Gutter EM. Famous people and genetic disorders: From monarchs to geniuses - A portrait of their genetic illnesses. Historical Review. Am J Med Genetics. 2003;118:187-96.
5. Beighton P. The Ehlers-Danlos syndromes. In: McKusicks heritable disorders of connective tissue. Whashinton: Mosby;1993. p.216-9.
6. Beighton P, De Paepe A, Steinmann B, Tsipouras P, Wenstrup RJ. Ehlers-Danlos syndromes: revised nosology. Villefranche, 1977. Ehlers-Danlos National Foundation (USA) and Ehlers-Danlos Support Group (UK). Am J Med Genet. 1998;77(1):31-7.
7. Bravo JF. Hereditary Disorders of the Connective Tissues (HDCT). A clinical study of 249 cases. Arth & Rheum. 2004;50(9)(Supl):S309.
8. Bravo JF. Síndrome de Hipermovilidad Articular. Cómo diferenciarlo de las otras alteraciones hereditarias de la fibra colágena. Reumatología. 2004;20(1):24-30.
9. Bravo J. Precauciones y posibles complicaciones quirúrgicas de las alteraciones hereditarias de la fibra colágena (AHFC). Rev Chilena de Cirugía. 57(6):516-22.
10. Weedon D. Piel Patología: Síndrome de Ehlers-Danlos. Madrid: Ed. Marban; 2002. p. 304-6.
11. Hussain A, Zeisberger S, Hubber P. Brittle cornea syndrome and its delineation from the kyphoscoliotic type of Ehlers-Danlos syndrome (EDS VI): report on 23 patients and review of the literature. Am J Med Genet. 2004 Jan 1;124A(1):28-34.
12. Fichard A, Chanut-Delalande H, Ruggiero F. [The Ehlers-Danlos syndrome: the extracellular matrix scaffold in question]. Med Sci (Paris). 2003 Apr;19(4):443-52.
13. Le Tallec H, Lassalle A, Khenioui H, Durufle A, Plassat R, Gallien P. Two cases of rehabilitation in Ehler-Danlos syndrome Ann Readapt Med Phys. 2006; 49(2):81-4
14. Anestesia subaracnoidea para una paciente con síndrome de Ehlers-Danlos tipo II . Fernández-García, A. Ramos-Zabala, C. Sanz-Baena B, R.Rev. Anestesiol Reanim. 2004;51:268-271.
15. Patel AB, Renge RL.Ehler-Danlos syndrome. Indian Pediatr. 2002;39(8):784-5.
16. Parikh F, Sivaramakrishnan A, Pai-dhungat JV .Type VI Ehler Danlos Syndrome. J Assoc Physicians India. 2004;52:631.
17. Sastry PS.Matrix metalloproteinase inhibitor therapy to prevent complications as well as therapy for Ehler-Danlos syndrome.Med Hypotheses. 2002; 59(3):314-5.
18. Keer R, Edwards-Fowler A, Mansi. Management of the hypermobile adult. In: Keer R, Grahame R, editors. Hypermobility Syndrome: Recognition and management for physiotherapists. Edinburgh, London, New York: Butterworth Heinemann. 2003;12:87-106.
19. Grahame R, Bird H. British consultant rheumatologists' perceptions about the hypermobility syndrome: A national survey. Source Rheumatology 2001;45:559-62.
20. Grahame R. Pain, distress and joint hyperlaxity. Joint, Bone, Spine 2000;67(3):157-63.
21. Gazit Y, Nahir AM, Grahame R, Jacob G. Dysautonomia in the hypermobility syndrome. American Journal of Medicine. 2003;115:33-40.
22. Sara E. Whitelaw.Ehlers-Danlos Syndrome, Classical Type: Case Management.Pediatr Nurs. 2003;29(6):423-6.
23. Grahame R, Bird HA, Child A. The British Society forRheumatology Special Interest Group on HeritableDisorders of Connective Tissue Criteria for the Benign Joint Hypermobility Syndrome. The revised(Brighton 1998) criteria for the diagnosis of BJHS. JRheumatol 2000;27:1777-79.
24. Maltz SB, Fantus RJ, Mellett MM, Kirby JP: Surgical complications of Ehlers-Danlos syndrome type IV: case report and review of the literature. J Trauma. 2001;51(2):387-90.
25. Persikov AV, Pillitteri RJ, Amin P, et al: Stability related bias in residues replacing glycines within the collagen triple helix in inherited connective tissue disorders. Hum Mutat. 2004; 24(4):330-7.
26. Ceccolini E, Schwartz R. Ehlers-Danlos Syndrome. 2006. [Citado 12 September 2007] disponible en: http://www.emedicine.com
27. Dalgleish R. The Human Collagen Mutation Database 1998. Nucleic Acids Research. 1998;26(1).
28. Nuytinck L, Freund M, Lagae L et al. Classical Ehlers-Danlos syndrome caused by a mutation in type I collagen; Am J Hum Genet. 2000;66:1398-402.
29. Shashikiran U, Rastogi A, Gupta RP, Sabir M. Ehler-Danlos syndrome type VI variant presenting with recurrent respiratory infections and responding to high dose vitamin C. J Assoc Physicians India. 1999; 47(5):554-5
30. Mao J, Bristow J. The Ehlers-Danlos Syndrome: Beyond collagens; J Clin Invest. 2001;107(9):1063-69.
31. Mao J, Bristow J. The Ehlers-Danlos Syndrome: Beyond collagens. J Clin Invest. 2001;107(9):1063-69.
Recibido: 8 de agosto de 2007.
Aprobado: 26 de noviembre de 2007.
George N. García Rodríguez. Agramonte 53, Apto. 3, entre Céspedes y Marti, Sancti Spiritus. Correo electrónico: mercury@ped.ssp.sld.cu


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}