










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Pediatría
versión On-line ISSN 1561-3119
Rev Cubana Pediatr v.81 n.4 Ciudad de la Habana sep.-dic. 2009
Clorhidrorrea congénita: primer reporte en Cuba
Congenital chlorhydria: first report in Cuba
Carlos Ramírez Pérez,I Gladys Abreu Sera,II Dinorah Mulet Batista,III José del Campo Avile,IV Miguel Peña Hernández V
I Especialista de I Grado en Gastroenterología. Asistente de Pediatría. Hospital Pediátrico Universitario de Holguín (HPUH). Holguín, Cuba.
II Especialista de I Grado en Gastroenterología. Asistente de Pediatría. Hospital Pediátrico Universitario de Holguín (HPUH). Holguín, Cuba.
III Especialista de I Grado en Laboratorio Clínico. Instructor de Laboratorio Clínico (HPUH). Holguín, Cuba.
IV Especialista de I Grado en Laboratorio Clínico. Instructor de Laboratorio Clínico (HPUH). Holguín, Cuba.
V Especialista de I Grado en Pediatría. Asistente de Pediatría (HPUH). Holguín, Cuba.
RESUMEN
En el presente trabajo se exponen los antecedentes prenatales, perinatales y posnatales de un lactante de 6 meses de edad, del sexo masculino, con clorhidrorrea congénita, así como el cuadro clínico, diagnóstico y tratamiento utilizado. Un elemento significativo lo constituyó la expulsión anal de abundante líquido no meconial desde las primeras horas de nacido, así como alcalosis metabólica grave y la presencia de desnutrición rápidamente progresiva. En los exámenes complementarios se constató hipocloremia de un 50 % con respecto a las cifras de referencia y un pH sanguíneo mayor de 7,50. El diagnóstico confirmatorio se obtuvo al comprobar concentraciones de cloro en heces fecales superiores a las de la suma de sodio y potasio. Se proponen indicadores diagnósticos prenatales entre los que tienen gran valor la consanguinidad, el polihidramnios y los resultados del ultrasonido en el tercer trimestre del embarazo. El diagnóstico precoz permitió establecer el tratamiento y evitó el frecuente desenlace fatal. Es el primer caso de esta enfermedad que se informa en el país.
Palabras clave: Clorhidrorrea congénita, alcalosis metabólica, hipocloremia.
ABSTRACT
In present paper are showed the prenatal, perinatal and postnatal backgrounds from a male breast-fed baby aged 6 months presenting with congenital chlorhydria, as well as the clinical picture, diagnosis and treatment applied. A significant element was the anal expulsion of non-meconium abundant fluid from the first hours of born, as well as a severe metabolic alkalosis and the presence of a quickly progressive malnutrition. In complementary examinations was confirmed a 50% hypochloremia regarding the reference figures and a blood pH over 7,50. Confirmatory diagnosis was achieved verifying the chlorine concentrations in feces higher to that of the sum of sodium and potassium. Prenatal diagnostic indicators are proposed those with higher value including the consanguinity, polyhydramnios and the US results during the third trimester of pregnancy. Early diagnosis allowed us to establish the treatment and to prevent a fatal outcome. This is the firs case of this entity reported in our country.
Key words: Congenital chlorhydria, metabolic alkalosis, hypochloremia.
INTRODUCCIÓN
La clorhidrorrea congénita (CHC) fue descrita por primera vez en 1945 por Gambler y Darrow,1-11 y es una enfermedad rara, autosómica recesiva, causada por mutaciones en el cromosoma 7, de comienzo prenatal, caracterizada por la presencia de diarrea secretoria con alto contenido de cloro. Es secundaria a la ausencia o disminución del intercambio activo de cloro y bicarbonato a nivel del íleon terminal y colon. Esto produce una importante pérdida de cloro, sodio y potasio en la materia fecal, con alcalosis hipoclorémica e hipopotasemia.1-6 Ante la sospecha clínica se debe realizar la determinación de cloro en la materia fecal, y su alta concentración es el criterio diagnóstico final. Ésta habitualmente excede los 90 mmol/L, con una media de 150 mmol/L. Luego de los 3 meses de edad, los valores por lo general superarán las sumas de las concentraciones de sodio y potasio fecales, cuyos valores medios suelen ser de 60 y 45 mmol/L, respectivamente.
En los casos de deshidratación grave y diarrea crónica, con depleción de electrolitos, el valor del cloro puede llegar a ser tan bajo como 40 mmol/L. Una vez corregida la deshidratación y alcanzado el equilibrio hidroelectrolítico, suele exceder los 90 mmol/L.7-11 La ausencia de cloruria es la regla. La muestra de materia fecal se debe obtener con un catéter rectal blando, para evitar la contaminación con orina.
La determinación del cloro en la materia fecal es la clave del diagnóstico: se observa una alta concentración. El tratamiento se basa en el reemplazo de las pérdidas fecales de cloro, sodio y agua, lo cual no evita la diarrea pero sí sus consecuencias secundarias,11 además de la utilización de medicamentos que disminuyan la secreción gástrica de cloro. El pronóstico depende del diagnóstico temprano, así como de una adecuada sustitución de electrolitos perdidos.1-3 El propósito de este trabajo fue presentar un caso de CHC, por tratarse de una causa no frecuente de diarrea acuosa congénita, de diagnóstico sencillo, que requiere un alto índice de sospecha, y además, establecer pautas para su detección temprana, inclusive antes del nacimiento.
PRESENTACIÓN DEL CASO
Paciente del sexo masculino, blanco, nacido a las 36 semanas de gestación mediante parto distócico (cesárea por preeclampsia materna), peso de 2000 g y antecedentes de consanguinidad en los padres (primos hermanos), quien fue valorado por los especialistas del servicio de gastroenterología a los18 días de nacido.
Antecedentes patológicos prenatales: polihidramnios y hallazgo ultrasonográfico de exceso de líquido intestinal en el feto, con sospecha de malformación congénita digestiva (se descartó horas después de nacido al comprobarse buen tránsito intestinal).
Antecedentes patológicos perinatales: expulsión de líquido anal no meconial en las primeras horas del nacimiento, antes del comienzo de la vía oral, lo cual se mantuvo durante varias semanas.
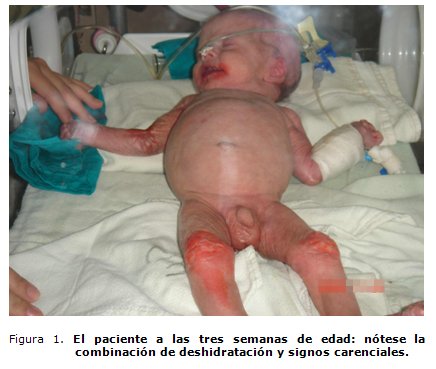
Antecedentes patológicos posnatales: deshidratación, desnutrición grave y progresiva; evidentes signos carenciales en la piel y mucosas (figura 1), alcalosis recidivante; en ocasiones con pH por encima de 7,60 y durante una descompensación llegó a 7,81; cloro en sangre disminuido en un 50 %; glicemia de 3,3 mmol/L.
El examen de rayos X ofreció los elementos siguientes: tórax normal, catéter umbilical bien ubicado, sonda nasogástrica en estómago y dilatación de asas intestinales.
Estudio del líquido intestinal: algunos leucocitos; algunos hematíes; Pandy positivo dos cruces.
El paciente siguió expulsando líquido anal de aspecto no meconial, luego de una semana de edad. Con el tratamiento el niño se recuperó y al hacer este informe tenía 6 meses de edad y se mantenía con buen estado general.
DISCUSIÓN
La CHC, a pesar de ser una entidad muy rara, presenta manifestaciones muy típicas y está entre las causas de diarrea grave del recién nacido, por lo que una vez que se ha descartado la inmadurez o déficit enzimático de disacaridasas, hay que sospecharla.
Con los resultados de los exámenes complementarios mencionados en la presentación, más la determinación de electrolitos en heces fecales y con el análisis clínico del caso, nos propusimos hacer un diagnóstico de certeza de acuerdo con los criterios mundialmente aceptados (figura 2).

En las series revisadas,1,11,12 el diagnóstico se ha realizado habitualmente luego de los 6 meses del nacimiento y basado en estudios radiológicos, así como en la frecuente alcalosis recidivante. Se sugiere, luego de estudiar este caso, un diagnóstico de sospecha prenatal, dado por la presencia de líquido intestinal excesivo en el feto (ecografía), más polihidramnios -reportado en la mayoría de las series revisadas-1,3,4,11,12 y consanguinidad.
Nuestra propuesta diagnóstica está basada en el hallazgo de líquido intestinal excesivo en el feto durante el tercer trimestre del embarazo. Este hecho constituyó un elemento bastante constante en los diferentes exámenes realizados. Además, el surgimiento de equipos de sonografía de mayor resolución posibilita este hallazgo. La imagen reportada es similar a la esperada en casos de estenosis congénita del intestino, donde lo predominante es la acumulación de líquido en algunos segmentos, más que la visualización del sitio exacto de estenosis.
Por tanto, en todo feto que presente un ultrasonido con las características descritas y que a estas se le sumen el polihidramnios o la consanguinidad (uno o ambos elementos), hay que tener en cuenta la CHC. En la propuesta de sospecha prenatal, le asignamos un mayor valor al hallazgo de líquido intraluminal, porque el polihidramnios es mucho más frecuente en la práctica obstétrica, ya sea asociado o no a otras entidades, por lo que su presencia solitaria es inespecífica, y no así en combinación con los elementos mencionados.
Como tercer indicador de sospecha señalamos la consanguinidad debido a que si es un trastorno autosómico recesivo, el problema tiene mayor probabilidad de expresarse cuando los progenitores portadores o enfermos están emparentados, a no ser que se trate de una mutación en el caso que se estudia. Al tratarse de una entidad sumamente rara y siguiendo los principios de la probabilidad, se infiere que no son abundantes las mutaciones en casos recientes, pues entonces habría mayor incidencia en la población en general. De esta forma se expresa el valor que tiene la consanguinidad como sospecha diagnóstica, mayor que en otros trastornos de igual patrón hereditario pero más frecuentes en la práctica médica (figura 3).

Existe un último elemento humoral de gran importancia y que es señalado por la mayor parte de la literatura consultada.1,2,4,11 Se trata de la hipocloremia, que en nuestro paciente llegó a ser de un 50 % en relación con las cifras normales de referencia. Si bien este indicador no es viable para la sospecha prenatal ni para el diagnóstico definitivo, sí tiene valor posnatal inmediato. Igual valor tiene la alcalosis metabólica, que estuvo presente desde los primeros días de vida pero que se malinterpretó en un momento determinado como alcalosis respiratoria. Inclusive, el ritmo respiratorio característico de este evento no se debe asociar con la respiración propia de algunos neonatos o recién nacidos inmaduros. Iniciamos el tratamiento antes de la confirmación diagnóstica pues los medicamentos utilizados son bastante inocuos y han conseguido resultados terapéuticos espectaculares.
El diagnóstico precoz (a las 3 semanas de nacido) permitió formular una solución rehidratante electrolítica de acuerdo con las necesidades específicas de esta enfermedad, que vinculado con la terapia con inhibidores de la bomba de protones que se encargan de reducir el cloro gástrico (principal fuente de este electrolito en el organismo), revertió el desfavorable estado del paciente y evitó el frecuente desenlace fatal. El pronóstico antes de imponer tratamiento era incierto pues, con solo 3 semanas de nacido, el neonato presentaba desnutrición extrema, deshidratación, necesidad de alimentación parenteral, flebitis, riesgo inminente de muerte, entre otras complicaciones, y signos carenciales en piel y mucosas.
REFERENCIAS BIBLIOGRÁFICAS
1. Holmberg C, Perheentupa J, Launiala K, Hallman N. Congenital chloride diarrhea. Clinical analysis of 21 Finnish patients. Arch Dis Child 1977;52:255-67.
2. Walker J-FD, Jan AJMF, Abel M. Congenital intestinal transport defects. En: Walker WA, Durie PR, Hamilton JR, Walker-Smith JA, Watkins JB, eds. Pediatric gastrointestinal disease. Pathophysiology, diagnosis, management. USA: Decker Inc.; 2000. Pp. 701-26.
3. Hoglund P, Holmberg C, Sherman P, Kere J. Distinct outcomes of chloride diarrhea in two siblings with identical genetic background of the disease: implications for early diagnosis and treatment. Gut 2001;48:724-727.
4. 4-García Garcia ML, Manzanares López-Manzanares J, Urruzuno Telleira P, Medina López C, Medina Benítez E. Clinical course and analysis of a case of congenital chloride diarrhea. An Esp Pediatr 1991;34:237-8.
5. Vermeylen D, Godart S, Moretto M, Janssen F, Bouton JM. Long-term follow up of a case of severe congenital chloride diarrhea. Eur J Pediatr 1988;147:649-52.
6. Field M. Intestinal ion transport and the pathophysiology of diarrhea. J Clin Invest 2003;111:931-43.
7. OMIM Center for Medical Genetics. Online Mendelian Inheritance in Man. Johns Hopkins University (Baltimore, MD) and National Center for Biotechnology Information, National Library of Medicine (Bethesda, MD). Available at: http://www.ncbi.nlm.nih.gov/omim
8. Kere J, Lohi H, Hoglund P. Genetic Disorders of Membrane Transport III. Congenital chloride diarrhea. Am J Physiol 1999;276:7-13.
9. Moseley RH, Hoglund P, Wu GD, Silberg DG, Haila S, de la Chapelle A, Holmberg C, Kere J. Downregulated in adenoma gene encodes a chloride transporter defective in congenital chloride diarrhea. Am J Physiol 1999;276:G185-92.
10. Lubani MM, Doudin KI, Sharda DC, Shaltout AA, al-Shab TS, Abdul Al YK, et al. Congenital chloride diarrhoea in Kuwaiti children. Eur J Pediatr 1989;148:333-6.
11. Contreras Mónica. Caso clínico. Acta Gastroenter Latinoam. 2005;35(2).
12. Benedetti L. Diarrea crónica en el lactante: Una causa más para pensar. 6ta Jornadas Nacionales de Médicos Residentes. Sociedad Argentina de Pediatría. 31 marzo al 2 de abril 2005, Buenos Aires.
Recibido: 27 de febrero de 2009.
Aprobado: 16 de junio de 2009.
Carlos Ramírez Pérez. Hospital Pediátrico Universitario «Octavio de la Concepción y de la Pedraja». Holguín, Cuba.
Correo electrónico: cmramirez@hpuh.hlg.sld.cu

