










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Investigaciones Biomédicas
versión On-line ISSN 1561-3011
Rev Cubana Invest Bioméd v.29 n.4 Ciudad de la Habana oct.-dic. 2010
TRABAJOS DE REVISIÓN
Mecanismos fisiopatológicos del desbalance glomérulo-tubular en la hipertensión arterial
Pathophysiological mechanisms of the lack of glomerulus-tubule balance in arterial high blood pressure
María Ofelia Barber FoxI; Katiana Galvizu DíazII; Aydelín Pérez RamosIII; María Ofelia Fox Pascual IV
IDoctora en Ciencias Médicas. Especialista de II Grado en Fisiología Normal y Patológica. Profesora Titular. FCM "Enrique Cabrera". La Habana, Cuba.
IIEspecialista de I Grado en Fisiología Normal y Patológica. Asistente. FCM "Salvador Allende". La Habana, Cuba.
IIIEspecialista de I Grado en Fisiología Normal y Patológica. Instructora. ICBP "Victoria de Girón". La Habana, Cuba.
IVDoctor en Ciencias Médicas. Profesora Titular. Profesora Consultante. ELAM. La Habana, Cuba.
RESUMEN
Las actuales tendencias e hipótesis para interpretar los mecanismos etiopatogénicos de la hipertensión arterial esencial, involucran al sistema renal como mecanismo preponderante en la regulación a largo plazo de la presión arterial y la existencia en él de algún fenómeno que puede conllevar a desbalance glomérulo-tubular, con preponderancia tubular Aunque el análisis de este último hecho no ha sido como tal abordado en la patogénesis del síndrome hipertensivo. Con el objetivo de interpretar el papel del desbalance glomérulo-tubular, con preponderancia tubular en la fisiopatología de la hipertensión arterial como fenómeno en el que confluyen múltiples mecanismos fisiopatológicos renales ya descritos, se revisaron estos últimos, de forma integrada y su relación causal con el desbalance glomérulo-tubular, con preponderancia tubular. La preponderancia tubular, punto común de los mecanismos que se discuten, favorece la disminución de la excreción fraccional de Na+, la retención hidrosalina y la elevación de la presión arterial.
Palabras clave: desbalance glomérulo tubular, hipertensión arterial y riñón, función tubular e hipertensión.
ABSTRACT
The current trends and hypotheses to know the etiopathogenesis mechanisms of the essential arterial high blood pressure involved the renal system as a prevailing mechanism in the long-term regulation of arterial pressure and the existence in it of some phenomenon that could lead to a glomerulus-tubule lack of balance with tubular preponderance. Although the analysis of this latter fact, has not been approached as such in pathogenesis of hypertensive syndrome. With the aim of to interpret the role of glomerulus-tubule lack of balance with tubular preponderance in pathophysiology of arterial high blood pressure as a phenomenon in which converging multiple renal pathophysiological mechanisms already described, these latter were reviewed in a integrated way and its causal relation with the above mentioned lack of balance with tubular preponderance. This preponderance, a common point of discussed mechanisms, favors the decrease of a fractional releasing of Na+, the hydrosaline retention and the raise of arterial pressure.
Key words: Tubular-glomerular lack of balance, arterial high blood pressure and kidney, tubular function and high blood pressure.
BALANCE GLOMÉRULO-TUBULAR
El riñón desempeña un importante papel en la regulación a largo plazo de la presión arterial (PA). Este órgano es el principal involucrado en la regulación del volumen del líquido extracelular y del sanguíneo, mediante el ajuste de su función a las variaciones de la ingestión de estos elementos, a través del mecanismo de diuresis y natriuresis por presión y, por tanto, de la excreción de sodio y agua. El ajuste de la excreción hidrosalina tiene lugar con la participación del balance existente entre la filtración glomerular y la reabsorción tubular (BGT), lo que resulta, junto a mecanismos neurohumorales, en el mantenimiento del balance entre los ingresos y egresos de sodio y agua en el organismo. Este fenómeno, consecuentemente, mantiene las cifras de PA dentro de límites fisiológicos.1
El BGT depende de dos mecanismos: la reabsorción proximal de sodio y agua regida por las fuerzas de Starling, que se establece entre los espacios intercelulares laterales a las células tubulares y el capilar peritubular; el segundo de estos mecanismos se relaciona con las cargas filtradas de glucosa y aminoácidos. Un mecanismo recientemente propuesto por Zhaopeng Du, consiste en la función mecanosensorial de las microvellosidades del borde en cepillo, ante el aumento del flujo tubular, lo cual aumenta la actividad del intercambiador Na+/H+ (NHE) en el proximal.2,3
Sin embargo, las alteraciones de las funciones de la filtración glomerular y reabsorción tubular, en el sentido de la preponderancia de la última, conllevarían a un rompimiento del BGT (DBGT-T) e interferirían con el mantenimiento de la homeostasis del Na+, conduciendo a retención hidrosalina y aumento de la PA.4
MECANISMOS FISIOPATOLÓGICOS DEL DBGT-T
Mecanismos que modifican la filtración glomerular
Hiperreactividad del sistema nervioso simpático. Según la teoría de Miasnikov, la tensión psíquica exagerada y mantenida provoca el agotamiento de los centros encefálicos de regulación vascular, lo que puede intensificar la actividad del sistema nervioso autónomo, especialmente del simpático (SNS) y así aumentar la actividad de este sobre los vasos del organismo y también del riñón.5
Se ha observado en ratas hipertensas espontáneas, una disminución de la sensibilidad del reflejo barorreceptor a los aumentos de la PA, debido a alteraciones encontradas en los núcleos del tractus solitario, lo que resulta en el mantenimiento de un tono simpático aumentado sobre el sistema cardiovascular y los vasos renales, provocando una vasoconstricción arteriolar aferente, que disminuye el flujo sanguíneo renal (FSR) y la filtración glomerular (IFG).6
Elevaciones de las concentraciones de catecolaminas. En algunos pacientes con HTA se han observado concentraciones plasmáticas elevadas de catecolaminas, fundamentalmente norepinefrina. Estos aumentos de catecolaminas en sangre están relacionados con elevaciones repetidas y exageradas de la PA por incrementos de la resistencia vascular (RVasc). Estas pueden también producir cambios anatómicos en los vasos sistémicos y renales. Sus efectos en las células musculares lisas vasculares (CMLV) tienen lugar a través de su interacción con receptores ß1-adrenérgicos (Fig. 1), activando los mecanismos intracelulares que se muestran en ella, y así disminuyen el FSR y la TFG.7-9
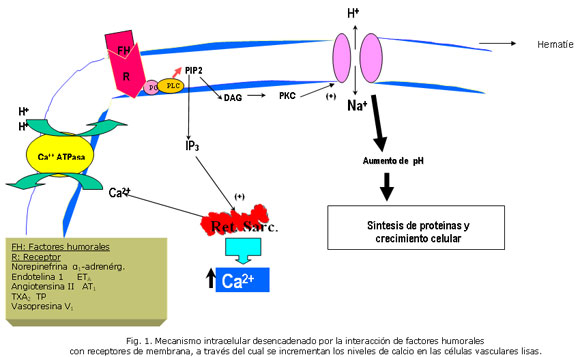
En la figura 1 se puede observar la vía del fosfatidil-inositol, la que se bifurca después de la hidrólisis del fosfatidilinositol 4,5 difosfato (PIP2), en la formación de dos segundos mensajeros, el diacilglicerol (DAG) y el Inositol trifosfato (IP3 ), cada uno de los cuales tiene mecanismos de acción diferentes, los CUALES conllevan a los incrementos intracelulares del pH y del Ca2+, respectivamente
Derivados del endotelio vascular: Entre los derivados endoteliales, con acciones vasoconstrictoras, se encuentra un conjunto de péptidos de 21 aminoácidos, denominados endotelinas 1, 2 y 3. La endotelina 1 (ET1) es el de mayor poder vasoconstrictor de todos.10
La ET1 interactúa con receptores ETA y ETB, estos se encuentran distribuidos de manera diferente en la pared vascular; el primero es predominante en las CMLV y el segundo en las células del endotelio vascular. La interacción de la ET1 con los receptores ETA, resulta en vasoconstricción y respuestas proliferativas de la pared vascular, mientras que su efecto a través de los receptores ETB, menos abundantes, es una vasodilatación, provocada mediante el estímulo de la liberación de óxido nítrico y prostaciclina por el endotelio.11
Los eventos intracelulares provocados por la unión de la ET1 con los receptores ETA en las CMLV, fundamentalmente sobre la arteriola aferente son aquellos que muestra la figura 1: la elevación de las concentraciones intracelulares de inositoltrifosfato (IP3), fenómeno que es regulado por la proteína G y mediado por la acción hidrolizante de la fosfolipasa C (FLC) sobre el fosfolípido de membrana 4,5-difosfato fosfatidilinositol. El incremento de IP3 trae como consecuencia la liberación de Ca2+ de sus sitios de almacenamiento intracelulares. Una mayor entrada de Ca2+ procedente del espacio extracelular es también estimulada por la ET1. La adición de estos dos efectos aumenta las concentraciones intracelulares de calcio y resulta en vasoconstricción.12
Una potente acción mitogénica posee la ET1, la cual involucra la acción de la proteína quinasa C, la tirosinquinasa, la mayor producción de factor de crecimiento derivado de las plaquetas (PDGF) y la elevación del pH intracelular por la activación del NHE, las que constituyen señales estimulantes de la proliferación y crecimiento celular. Este fenómeno es responsable de la hipertrofia de la pared vascular (HTV) observada bajo el efecto de altas concentraciones plasmáticas de ET1.13
Por tanto el aumento mantenido de la RVasc en el organismo y en el riñón, determinado por niveles plasmáticos o locales elevados de ET1, es consecuencia de vasoconstricción e HTV. Ambos fenómenos conllevan, en el riñón, a disminuciones del flujo sanguíneo y la filtración glomerular.14
La ET1, también pudiera interferir con la IFG, disminuyendo el coeficiente de filtración (Kf), debido a que provoca proliferación de las células mesangiales, el depósito de colágeno tipo IV, fibronectina y laminina en la lámina basal glomerular y, por otro lado, la contracción del mesangio. Estos procesos determinan un aumento del grosor de la barrera de filtración y una disminución de su área, respectivamente.15,16
El tromboxano A2 (TXA2), una variedad de prostanoide con acción vasoconstrictora sobre la arteriola aferente fundamentalmente, incrementa los niveles intracelulares de Ca2+ por la misma vía intracelular de la ET1, a partir de la interacción del TXA2 con receptores específicos (TP) acoplados a la proteína G (Fig. 1). En el riñón, el TXA2 además estimula la contracción mesangial y la proliferación de las células de esta estructura. Como resultado de ambas acciones disminuye el FSR y la filtración en el glomérulo.17,18
La producción de especies reactivas del oxígeno por las células endoteliales, ante estímulos como la angiotensina II y fuerzas mecánicas ejercidas sobre el endotelio, adiciona sus efectos vasoconstrictores aferentes a los factores humorales ya analizados. Fundamentalmente el O2- y el OH. producen potentes constricciones vasculares en los vasos renales, por acciones directas sobre la fibras musculares lisas y por vía indirecta mediando las acciones de sustancias vasoconstrictoras como la angiotensina II o a través de la inactivación del óxido nítrico que produce el O2-.19
Por otra parte, el óxido nítrico (ON) es el principal exponente de los derivados endoteliales, que determinan vasodilatación. La figura 2 muestra la unión de este al grupo hemo de la guanilatociclasa en las CMLV, la cual eleva los niveles citoplasmáticos de GMPc y conlleva al incremento en la actividad de la bomba Ca2+ ATPasa y del intercambiador Na+/Ca2+. Además el GMPc, conduce a la inactivación de los canales de Ca2+ dependientes de voltaje. La resultante de todos estos efectos es la disminución del Ca2+ citosólico y vasodilatación.20,21
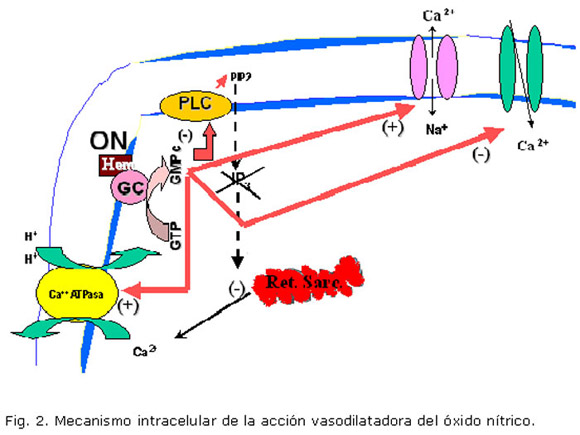
Las acciones del ON son de gran importancia en el mantenimiento de la función renal, la cual resulta severamente afectada cuando se produce inhibición prolongada de su síntesis o liberación, debido a la vasoconstricción sostenida y al daño glomerular a que esto conduce. Como puede deducirse de lo anterior, la consecuencia de una menor síntesis y liberación de ON en la vasculatura renal es una menor IFG.22
Otro factor relajante de la musculatura vascular, sintetizado predominantemente en las células del endotelio vascular renal; lo constituye uno de los metabolitos del ácido araquidónico (AA): la prostaciclina (PGI2), cuyos efectos vasodilatadores mantienen el FSR y la IFG aun en circunstancias como el aumento de las concentraciones de AII, vasopresina y noradrenalina. Su mecanismo de acción radica en la elevación de los niveles citoplasmáticos de AMPc, en las CMLV.23
También se ha observado que la PGI2 influye sobre la función glomerular, contrarrestando la acción de hormonas que provocan contracción del mesangio, como la AII e inhibe la proliferación de las células mesangiales ante la aparición de estímulos mitogénicos.24
Disfunción endotelial. La disfunción endotelial es un estado del endotelio vascular en el que se establecen alteraciones metabólicas y de la expresión génica de los agentes vasoactivos derivados del endotelio, rompiéndose el equilibrio existente entre la producción de agentes vasoconstrictores y vasodilatadores.25
En algunos modelos de hipertensión experimental y en el humano hipertenso, ha sido descrita la existencia de disfunción, que trae como consecuencia el predominio de los factores constrictores, debido a lo cual se produce una vasoconstricción mantenida. Múltiples investigadores han fundamentado que este fenómeno puede conducir a la disminución de la excreción de agua y sal y elevar la PA. La disfunción endotelial está frecuentemente asociada a mayor producción de AII, de radicales libres, mayor fuerza de rozamiento sobre la pared endotelial, al igual que mayor fuerza de cizallamiento.26
En la actualidad múltiples investigadores concuerdan con la idea de que la disfunción endotelial es consecuencia y no causa de la hipertensión.27,28
Derivados del ácido araquidónico: El AA es un ácido graso polinsaturado asociado a la membrana plasmática, formado a partir del ácido linoleico; su metabolismo en el riñón se realiza a través de 3 vías enzimáticas: cicloxigenasa, lipoxigenasa y citocromo P-450. Mediante la primera vía tiene lugar la formación de prostaglandinas (PGs) y TXA2, siendo esta vía la predominante para el metabolismo del AA en el riñón.
Las PGs y el TXA2 poseen acciones en el músculo liso vascular, en las células mesangiales, en el transporte de sal y agua y sobre la liberación de renina en el riñón.29,30
El 80 % de las PGs que se producen en el riñón lo constituyen la prostaglandina E2 (PGE2) y la prostaciclina (PGI2). La PGI2 posee efectos sobre la función glomerular que fueron discutidos en el acápite de derivados endoteliales. La PGE2 es sintetizada en diferentes sitios del riñón como el aparato yuxtaglomerular, el mesangio, el túbulo distal, conducto colector y en el intersticio renal. La PGE2 tiene un efecto vasodilatador y relajante del mesangio.31,32
La vía a través de la citocromo P450 es una vía metabólica del AA menos abundante en el riñón, da origen a los ácidos epoxyeicosatrienoico (EET) e hidroxieicosatetraenoico (HETE), los cuales pudieran provocar vasodilatación y vasoconstricción, respectivamente, determinando variaciones de la IFG. En algunos modelos de hipertensión como en las SHR, el inducido por infusión de angiotensina II y otros, se ha comprobado un aumento de producción de 20-HETE, lo que se responsabilizó con disminución de la IFG. Sin embargo en las ratas Dahl se ha asociado una disminución de este mismo metabolito con aumento de reabsorción tubular de Na+ e hipertensión.33
Sistema calicreína-cinina: El sistema calicreína-cinina participa en la regulación de la función renal. Los principales componentes de este sistema son la calicreína, el cininógeno, las cininas y las cininasas. Existen en el organismo 2 tipos diferentes de calicreínas: la plasmática y la tisular, las cuales difieren en sus pesos moleculares, características inmunológicas y físicas. La primera de ellas, activa a este sistema en el plasma y la segunda se encuentra predominantemente en el riñón, glándulas salivares, páncreas, intestino y también en el plasma. La bradicinina constituye el péptido vasoactivo básico de este sistema en el riñón.
Las cininas formadas en el tejido renal poseen una potente acción vasodilatadora sobre las arteriolas aferentes y eferentes directa e indirectamente, esto último por estimular la liberación de PGE2 y de ON, y así determinan incrementos de FSR y IFG. Finalmente las cininas se convierten en productos de degradación por la acción de las cininasas I y II. La cininasa II no es más que la enzima convertidora de angiotensina I en II (ECA), punto en que convergen ambos sistemas de péptidos vasoactivos renales.34
La importancia de la acción en el riñón de las cininas y su repercusión sobre la regulación de la PA ha sido planteada por Katori M y otros sobre la base de estudios realizados en ratas Brown-Norway Katholiek, congénitamente deficientes de cininógeno y también en el receptor B2 para la bradicinina, de ratones knockout. Estos autores han fundamentado teóricamente que un defecto congénito en la secreción de calicreína pudiera desempeñar un papel importante en el desarrollo de la HTA sal-sensible.35
Insulina: Se ha observado la existencia de resistencia tisular a la insulina (RTI) y aumento de las concentraciones de esta hormona, en el plasma de pacientes hipertensos esenciales y en modelos experimentales de hipertensión. También se ha demostrado la estrecha relación existente entre la hiperinsulinemia y la elevación de la PA, así como la importancia de la participación de la RTI en el desarrollo de insuficiencia renal en el paciente hipertenso esencial.36
La insulina activa al SNS y el desarrollo de (HTV), contribuyendo al remodelado vascular observado en hipertensos esenciales, el que aumenta la RVasc en el organismo en general y en los vasos renales. Esto último conllevaría junto a otros factores de los ya analizados, a un menor FSR, con la consecuente caída de la IFG.37
Vasopresina: La vasopresina (ADH) se libera en la hipófisis posterior frente a estímulos como: la mayor osmolaridad efectiva del líquido extracelular, la caída de la PA y del volumen sanguíneo, la acción de la AII, entre otros. La ADH provoca vasoconstricción sistémica y renal, por lo que disminuye el FSR y la IFG.38,39
La ADH interactúa con 2 tipos de receptores: V1 y V2, presentes en las CMLV y en los túbulos colectores, respectivamente. La hormona provoca vasoconstricción, a través del aumento del Ca2+ intracelular, por su unión a receptores del tipo V1. También incrementa la actividad del NHE en esas células, favoreciendo así la (HTV), el incremento de la resistencia sistémica y renal, y una menor IFG.40
Sistema renina-angiotensina (SRA): Los principales componentes de este sistema son la renina, el angiotensinógeno, la angiotensina I, la angiotensina II, la ECA, la aminopeptidasa A y los fragmentos peptídicos derivados de las sucesivas hidrólisis que ocurren en esta cascada enzimática. La AII es el péptido vasoactivo más potente de todo este sistema.
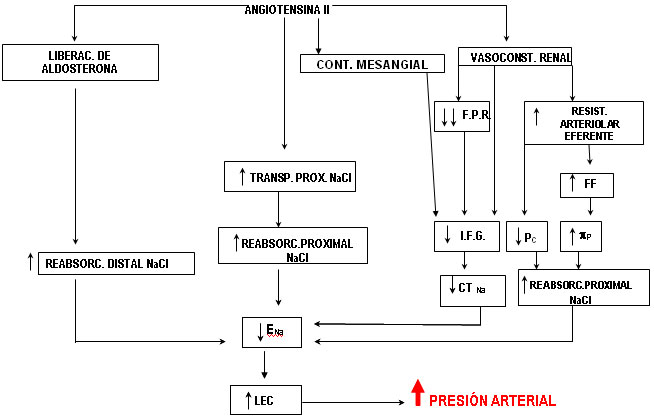
Múltiples acciones sistémicas relacionadas con la regulación de la PA, han sido descritas para la AII. En la figura 3 se han ilustrado aquellas acciones de la angiotensina II que afectan directamente la función renal, para simplificar los mecanismos de este sistema que, actuando sobre el riñón, pueden conllevar a la elevación de la PA. Como se observa, la AII provoca modificaciones del FSR, la IFG y el transporte tubular de Na+. Esta causa vasoconstricción fundamentalmente en la arteriola eferente, por lo que disminuye el FSR y mantiene la IFG ante la caída del primero, fenómeno que conlleva a una elevación de la fracción de filtración. Este péptido interactúa con receptores de tipo AT1 y AT2. La disminución de la excreción renal y la elevación de la PA son consecuencia de su unión a los del primer tipo (figura 1).41
A pesar de que el efecto inicial de la AII sobre la vasculatura renal, tiende a mantener la filtración glomerular dentro de valores normales (120 mL/min), con el paso del tiempo la caída de la IFG sobreviene, como consecuencia de la disminución mantenida del FSR. A lo anterior se adiciona la contracción del mesangio y el depósito de fibronectina, laminina y colágeno tipo IV en la membrana basal glomerular. Todo este conjunto de acciones contribuyen, en diferentes plazos de tiempo, a la reducción de la filtración, por disminución del área de la superficie de difusión, por aumento del grosor de la barrera filtrante y disminución de la conductividad hidráulica de estas estructuras.42
Además, la AII favorece el remodelado vascular, actuando a través de los receptores AT1, ya que induce la expresión de factores angiogénicos y de crecimiento, como son el factor básico de crecimiento de los fibroblastos (bFGF), el factor de crecimiento transformador beta (TGF-ß) y el PDGF.43
En estudios recientes se ha observado que la AII puede provocar vasoconstricción y disminución del FSR, a consecuencia de la activación de una oxidasa (NADP-H-dependiente) asociada a la membrana de las CMLV, la cual determina una mayor producción de O-2. Este último inactiva al ON convirtiéndolo en nitrito. Bajo estas condiciones gran parte del ON liberado del endotelio de los vasos renales, se inactiva y pierde su acción vasodilatadora, predominando los factores vasoconstrictores.44
Factor similar a la ouabaína (OLF): Hormona liberada de la corteza de la glándula suprarrenal, aunque existen niveles tisulares de la misma en el hipotálamo, la hipófisis, el corazón y el riñón. Elevaciones plasmáticas de OLF, se han observado en pacientes con HTA, cuando una expansión de volumen tiene lugar. En los vasos sistémicos y renales, el OLF aumenta la RVasc por los aumentos citosólicos de Ca2+ y de Na+ en las CMLV, debido a la inhibición de la bomba Na+/K+ ATPasa y al incremento del influjo de Ca2+ por el intercambiador Na+/Ca2+ tipo 1.45
Alteraciones en las membranas plasmáticas de las fibras musculares lisas vasculares: Se han observado diferentes alteraciones de la membrana plasmática de las CMVL en los vasos sanguíneos de pacientes hipertensos esenciales y en ratas hipertensas espontáneas, que conllevan al incremento de la RVasc.46,47 Este fenómeno en la vasculatura renal conduce a la disminución de la IFG. Estas alteraciones son:
- Disminución de la actividad de la bomba Ca2+ ATPasa
- Disminución de la actividad de la bomba Na+/K+ ATPasa
- Aumento del influjo de Ca2+ a través del intercambiador Na+/Ca2+ tipo 1
- Aumento de la permeabilidad de la membrana para el Na+
Mecanismos que modifican la reabsorción tubular de Na+:
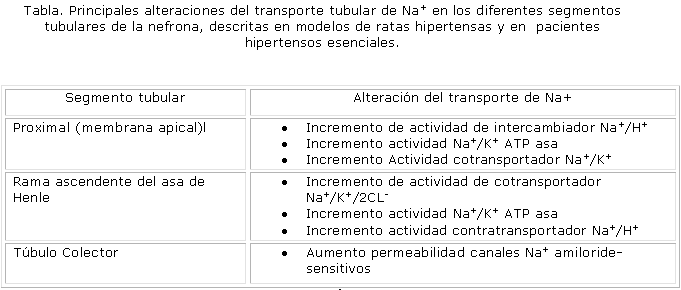
Aumento del transporte de sodio por modificaciones morfofuncionales en las células tubulares: En variadas ocasiones se ha planteado que la menor eliminación renal de Na+ responde, en varios modelos de ratas y en una parte de los pacientes hipertensos, a mayor transporte tubular reabsortivo de este ion, el cual suele ser causado por alteraciones genéticas o variaciones en la producción de determinados autacoides renales. En la tabla aparecen relacionadas las principales alteraciones del transporte tubular de Na+ que han sido descritas en los diferentes segmentos tubulares de la nefrona (tabla).
La menor liberación de ON en el riñón, una disfunción de los receptores dopaminérgicos D1 en el túbulo proximal, una mutación en el gen que codifica para la á aducina, una proteína del citoesqueleto importante en el transporte transmembrana de Na+ o la síntesis renal disminuida de 20-HETE; conducen a una mayor actividad de la bomba Na+/K+ ATPasa en la membrana basolateral de las células del túbulo proximal. La influencia de los 3 primeros factores, también puede provocar el incremento de la actividad NHE en este mismo segmento, fenómeno que también se observa bajo la acción de la ET1, sobre todo cuando sus concentraciones en la corteza renal se elevan.48
Por otra parte en la RGAAH, la disminución de la síntesis renal de 20-HETE y de ON, y el polimorfismo de la á aducina, son causantes de una mayor actividad del cotransportador Na+/K+/2Cl-, a lo cual se añade que estos 2 últimos factores estimulan la actividad de la bomba Na+/K+ ATPasa en este segmento. Mientras la disminución de la síntesis de ON también estimula el NHE, en cambio en el túbulo colector, la disminución de los niveles de ON conduce al aumento selectivo de la permeabilidad de los canales amiloride sensitivos para el Na+, situados en la membrana apical de las células de este segmento.49,50
Es de destacar la importancia que representa para la homeostasis del Na+ y para el volumen sanguíneo corporal, la incidencia del mayor número de los factores que se han mencionado, en aquellos sitios de la nefrona donde tienen lugar los mayores porcentajes de transportes reabsortivos de este ion: túbulo proximal (65 %) y RGAAH (25 %), lo que trae como consecuencia gran reabsorción de este y retención hidrosalina.
Insulina: En los túbulos renales, la insulina estimula la reabsorción de Na+ en la nefrona distal (RGAAH y túbulo distal) aunque se ha reportado una acción similar en el segmento tubular proximal en conejos. Esta hormona estimula la actividad del intercambiador Na+/H+ (NHE3), aumentando así el transporte reabsortivo de sodio y agua en los segmentos tubulares donde ejerce sus efectos.51
La razón por la cual las acciones de la insulina sobre los túbulos renales se intensifican en la hipertensión, ha sido postulada por Secchi, que plantea una conservación de la sensibilidad a esta hormona en las células tubulares y de su efecto antinatriurético.52
Angiotensina II: La AII estimula directamente la reabsorción de sodio en los túbulos renales (túbulo proximal y en la RGAAH) donde, interactuando con los receptores de tipo AT1, aumenta la actividad de la bomba Na+/K+ ATPasa y del Na+/HCO3- cotransportador en la membrana basolateral, y del NHE, en la zona apical de estas células. Además este péptido aumenta la permeabilidad de los canales epiteliales de Na+ en el conducto colector cortical, por esta misma vía. Adicionalmente desarrolla esta función, por la estimulación de la liberación de aldosterona como ya es bien conocido.41
Actividad del SNS: La inervación simpática es amplia en el riñón, los nervios simpáticos se encuentran en la vasculatura y en los distintos segmentos nefronales, con excepción de los conductos colectores. Generalmente el neurotransmisor presente es la norepinefrina y los receptores son á1-adrenérgicos. Además, existen receptores á2-adrenérgicos que no forman parte de las uniones sinápticas e interactúan con las catecolaminas circulantes, ambas clases de receptores se encuentran en el túbulo proximal y el último de ellos en los segmentos distales.
Estudios realizados en conejos y en ratas, han demostrado que la estimulación del SNS incrementa directamente el transporte tubular de sodio, por medio de la activación de la bomba Na+/K+ ATPasa y del NHE en el túbulo proximal, a través de receptores á1-adrenérgicos y á2-adrenérgicos.53
Factor similar a la ouabaína (OLF): Además de sus efectos sobre la vasculatura renal, esta hormona aumenta la expresión y actividad de la Na+/K+ ATPasa en los túbulos, lo que favorece la reabsorción tubular.54
Alteraciones genéticas relacionadas con el transporte tubular de Na+: Diversas alteraciones genéticas (monogénicas o polimorfismos) han sido encontradas en pacientes hipertensos o en modelos experimentales de hipertensión:55
Hipertensión monogénica: Aldosteronismo remediable por glucocorticoides: La hipertensión es producto de que el gen quimérico sitúa a la aldosterona sintetasa bajo el control de la ACTH.
Exceso aparente de mineralocorticoides: Mutación del gen que codifica para la 11b-hidroxiesteroide deshidrogenada que convierte al cortisol en cortisona, impidiendo que el primero interactúe con los receptores para la aldosterona. Con esta mutación presente, el cortisol ocupa el lugar de la aldosterona y se desarrolla la hipertensión por expansión de volumen, debido a que esta interacción entre el cortisol y los receptores de mineralocorticoide tiene como resultante el aumento de la actividad de los canales epiteliales de Na+.
Síndrome de Liddle: Mutaciones de los genes que codifican para las subunidad â o ã del canal epitelial de Na+, lo que aumenta el número de canales de este tipo en los túbulos, además de que este permanece inapropiadamente permeable, a pesar de una gran ingestión de sal.
Mutación activadora del receptor de mineralocorticoide: Esta provoca el funcionamiento de este receptor independiente de la acción de la aldosterona.
Hipertensión primaria: Polimorfismos de genes responsables de la expresión de componentes del SRA: polimorfismo del gen que codifica al angiotensinógeno (M235T), polimorfismo del gen de sintetasa de aldosterona (T344C), polimorfismo del gen de la enzima convertidora de angiotensina I (ECA D/I), polimorfismos del gen de AT1 receptores (AII66C).
Polimorfismo de la á aducina: Bianchi y otros observaron que este se asocia a incrementos del transporte de Na+ en los túbulos renales y en los eritrocitos de ratas MHS. Esta mutación está en relación con aumentos de la actividad del cotransportador Na+/K+/2Cl- en la membrana de las células de la RGAAH y ha sido también relacionada con incremento de la actividad de la bomba Na+/K+ en la membrana basolateral de este segmento y en las células del túbulo proximal.
Mutación del gen de la subunidad â del canal epitelial del sodio: Baker y su grupo describieron la existencia de la mutación T594M en la subunidad â del canal epitelial de Na+ en hipertensos esenciales de la raza negra, lo que tiene como consecuencia funcional, una mayor reabsorción de este ion.
En resumen, la resultante de la acción en el riñón de todos los mecanismos analizados, es la disminución de la IFG y/o el aumento de la reabsorción tubular, teniendo como punto común la preponderancia de la función tubular y el establecimiento de un DBGT-T, lo que favorece la incapacidad renal para eliminar Na+ y agua, la retención hidrosalina, la expansión del volumen sanguíneo y la elevación de la presión arterial, fenómeno que, de mantenerse, conllevaría al desarrollo de la HTA.
En correspondencia con lo hasta aquí expuesto, en investigaciones precedentes nosotros establecimos un DBGT-T, a partir de generar una hipertrofia tubular proximal por medio de inhibidores del SRA y en ellas se demostró que este fenómeno fue causa del desarrollo de HTA en ratas, que antes de los experimentos eran normotensas.56-61
REFERENCIAS BIBLIOGRÁFICAS
1. Guyton AC. Blood pressure control: Special role of the kidneys and body fluids. Science. 1991;252:1813-16.
2. Koeppen BM, Stanton BA. Renal Physiology: Renal transport mechanisms. 3th ed. Missouri: Mosby; 2001. p. 49-73.
3. Zhaopeng D, Yi D, QingShang Y, Weinstein AM., Weinbaum S, Wang T. Mechanosensory function of microvilli of the kidney proximal tubule. Proc Natl Acad Sci USA. 2004 August;101(35):13068-73.
4. Cervenka L, Simova M, Maly J, Heller J. Role of the kidney in long-term regulation of blood pressure and the development of hypertension. Cesk Fysiol. 2000 Aug;49(3):116-33.
5. Hernández A. Fisiopatología de la HTA esencial. Modelo Instructivo. 1994 [en línea] Disponible en: http://www.sld.cu/libros/hiperten/referencias.html. [Consultado: 22 Febrero 2009].
6. De Quattro V, Feng M. The sympathetic nervous system: the muse of primary hypertension. J Hum Hypertens. 2002 Mar;16(Suppl 1):S64-69.
7. Jacobs MC, Lenders JW, Willemsen JJ, Thien T. Adrenomedullary secretion of epinephrine is increased in mild essential hypertension. Hypertension. 1997;29:1303-08.
8. Miranda-Ferreira R, Pascual R, Diego A, Caricati-Neto A, Gandía L, Jurkiewicz A, et al. Single-vesicle catecholamine release has greater quantal content and faster kinetics in chromaffin cells from hypertensive, as compared with normotensive rats. JPET. February 2008;324(2):685-93.
9. Erami C, Zhang H, Tanoue A, Tsujimoto G, Thomas S, Faber J. Adrenergic catecholamine trophic activity contributes to flow-mediated arterial remodeling. Am J Physiol Heart Circ Physiol. 2005; 289:H744-53.
10. Karin I. El sistema renina angiotensina, estrés oxidativo y endotelina en el desarrollo de la hipertensión arterial [en línea]. Minnesota, USA. Disponible en: http://www.sac.org.ar/Publicaciones/boletin/1/sistemareninaangiotensina.pdf. [Consultado: 6 diciembre 2007].
11. Davenport AP, Battistini B. Classification of endothelin receptors and antagonists in clinical development. Clinical Science. 2002;103(Suppl 48):1S3S.
12. Schroede AC, Imig JD, LeBlanc EA. Endothelin-mediated calcium signaling in preglomerular smooth muscle cells. Hypertension. 2000;35:280.
13. Orlov SN. Na+-H+ exchanger as a target for intervention in cardiovascular remodelling. J Hypertens. 2003 Aug;21(8):1463-65.
14. Ozawa Y, Hasegawa T, Tsuchiya K, Yoshizumi M, Tamaki T. Effect of endothelin-1 (1-31) on the renal resistance vessels. The Journal of Medical Investigation. 2003 February;50(1):2.
15. Sorokin A, Foschi M, Dunn MJ. Endothelin signalling and regulation of protein kinases in glomerular mesangial cells. Clinical Science. 2002;103(Suppl 48):132S136S.
16. Sorokin A, Kohan D. Physiology and pathology of endothelin-1 in renal mesangium. Am J Physiol Renal Physiol. 2003;285:F579-89.
17. Wang D, Chabrashvili T, Wilcox CS. Enhanced contractility of renal afferent arterioles from angiotensin-infused rabbits: roles of oxidative stress, thromboxane prostanoid receptors, and endothelium. Circ Res. June 2004;94(11):1436-42.
18. Schmitz PG, Zhang K, Dalal R. Eicosapentaenoic acid suppresses PDGF-induced DNA synthesis in rat mesangial cells: involvement of thromboxane A2. Kidney Int. 2000 Mar;57(3):1041-51.
19. Schnackenberg CG. Physiological and pathophysiological roles of oxygen radicals in the renal microvasculature. Am J Physiol Regul Integr Comp Physiol. 2002;282(2):R335-42.
20. Llorens S, Jordán J, Nava E. The nitric oxide pathway in the cardiovascular system. J Physiol Biochem. 2002;58(3):179-88.
21. Kramp RA, Fourmanoir P, Ladrière L, Joly E, Gerbaux C, Hajjam A, et al. Effects of Ca2+ channel activity on renal hemodynamics during acute attenuation of NO synthesis in the rat. Am J Physiol Renal Physiol. 2000 April;278(4):F561-69.
22. Cachofeiro V, Fortepiani LA, Navarro-Cid J. Renal dysfunction after chronic blockade of nitric oxide synthesis. Antioxid Redox Signal. 2002 Dec;4(6):885-91.
23. Achata J. Secreción de las PGs. Mecanismo de acción de las prostaglandinas [en línea]. En: El riñón y la hipertensión arterial: Causa o consecuencia. Disponible en: URL: http://www.s.com/trabajos7/pros/pros.shtml. [Consultado:12 Marzo 2009].
24. Nasrallah R, Landry A, Scholey JW, Hébert RL. Characterization of the PGI2/IP system in cultured rat mesangial cells. Prostaglandins, leukotrienes and essential fatty Acids. 2004;70(5):455-64.
25. Fasce E. Disfunción endotelial e hipertensión. [en línea]. Hipertensión. Boletín Oficial de la Sociedad Chilena de Hipertensión. 1998;l7(1). Disponible en: http://www.udec.cl/~ofem/revista/revista03/artic4.html [Consultado: 20 Mayo 2006].
26. Gomazkov OA. The molecular and physiological aspects of endothelial dysfunction. The role of endogenous chemical regulators. Usp Fiziol Nauk. 2000 Oct-Dec;31(4):48-62.
27. Paniagua OA, Bryant MB, Panza JA. Transient hypertension directly impairs endothelium-dependent vasodilation of the human microvasculature. Hypertension. 2000;36:941.
28. Stankevicius E, Martinez AC, Mulvany MJ, Simonsen Ulf. Blunted acetylcholine relaxation and nitric oxide release in arteries from renal hypertensive rats. Journal of Hypertension. 2002;20(8):1571-79.
29. Imig JD. Eicosanoid regulation of the renal vasculature. Am J Physiol: Renal Physiol. 2000;279:F965-81.
30. Boron WF, Boulpaep EL. Glomerular filtration and renal blood flow [en línea] en: Medical Physiology. Disponible en: http://www3.us.elsevierhealth.com/MERLIN/BandB/supplements /Chapter_33.html [Consultado: 11 Junio 2008].
31. Fuson AL, Komlosi P, Unlap TM, Bell PD, Peti-Peterdi J. Immunolocalization of a microsomal prostaglandin E syntethase in rabbit kidney [en línea]. Am J Physiol: Renal Physiol. 2003 May; Disponible en: www.ajprenal.physiology.org/papbyrecent.shtml. [Consultado: 16 Diciembre 2004].
32. Hui-Fang Cheng; Raymond CH. Cyclooxygenases, the kidney, and hypertension. Hypertension. 2004;43:525.
33. Sarkis A, Lopez B, Roman RJ. Role of 20-hydroxyeicosatetraenoic acid and epoxyeicosatrienoic acids in hypertension. Curr Opin Nephrol Hypertens. 2004 Mar;13(2):205-14.
34. Imig JD. ACE Inhibition and bradykinin-mediated renal vascular responses. Hypertension. 2004;43:533.
35. Katori M, Majima M. The renal kallikrein-kinin system: its role as a safety valve for excess sodium intake, and its attenuation as a possible etiologic factor in salt-sensitive hypertension. Critical Reviews in Clinical Laboratory Sciences. 2003;40(1):43-115.
36. Kincaid-Smith P. Obesity and the insulin resistance syndrome play a major role in end-stage renal failure attributed to hypertension and labelled `hypertensive nephrosclerosis'. Journal of Hypertension. 2004;22(6):1051-55.
37. Andronico G, Ferraro-Mortellaro R, Mangano MT, Rome M, Raspanti F, Pinto A, et al. Insulin resistance and glomerular hemodynamics in essential hypertension. Kidney Int. 2002 Sep;62(3):1005-09.
38. Hormones of the pituitary [en línea]. Hormones of the pituitary 2003. Disponible en: http://www.users.rcn.com/jkimball.ma.ultranet/BiologyPages/P/Pituitary.html. [Consultado: 20 junio 2006].
39. Christiansen RE, Roald AB, Gjerstad C, Tenstad O, Iversen BM. Renal hemodynamics in young and old spontaneously hypertensive rats during intrarenal infusion of arginine vasopressin. Kidney Blood Press Res. 2001;24(3):176-84.
40. Henderson KK, Byron KL. Vasopressin-induced vasoconstriction: two concentration-dependent signaling pathways. J Appl Physiol. 2007;102:1402-09.
41. Kobori H, Nangaku M, Navar G, Nishiyama A. The intrarenal renin-angiotensin system: from physiology to the pathobiology of hypertension and kidney disease. Pharmacological Reviews. September 2007;59(3):251-87.
42. Chen S, Lee JS, Iglesias-de la Cruz MC. Angiotensin II stimulates á3(IV) collagen production in mouse podocytes via TGF-ß and VEGF signalling: implications for diabetic glomerulopathy. Nephrol Dial Transplant. 2005;20(7):1320-28.
43. Sun Y. The renin-angiotensin-aldosterone system and vascular remodeling. Congest Heart Fail. 2002;8(1):11-16.
44. López B, Salom MG, Arregui B, Valero F, Fenoy FJ. Role of superoxide in modulating the renal effects of angiotensin II. Hypertension. 2003;42(6):1150-6.
45. Blaustein MP, Zhang J, Chen L, Hamilton BP. How does salt retention raise blood pressure? Am J Physiol Regul Integr Comp Physiol. 2006;290:R514-23.
46. Vershinina AM, Gapon LI, Shurkevich NP, Krasnoperova IF, Petelina TI, Krinochkin DV. Role of cell membrane abnormalities in the etiology of arterial hypertension. Ter Arkh. 1998;70(12):24-8.
47. Iwamoto T. Vascular Na+/Ca2+ exchanger: implications for the pathogenesis and therapy of salt-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2006;290:R536-45.
48. Cusi D. Genetic renal mechanisms of hypertension. Curr Opin Nephrol Hypertens. 1997;6:192-204.
49. Orlov SN, Adragna NC, Adarichev VA, Hamet P. Genetic and biochemical determinants of abnormal monovalent ion transport in primary hypertension. Am J Physiol Cell Physiol. 1999;276:C511-36.
50. O'Shaughnessy KM, Karet FE. Salt handling and hypertension. J Clin Invest. 2004;113:1075-81.
51. Klisic J, Chang Hu M, Nief V, Reyes L, Fuster D, Moe OW, et al. Insulin activates Na+/H+ exchanger 3: biphasic response and glucocorticoid dependence. Am J Physiol Renal Physiol. 2002;283:F532-39.
52. Sechi LA. Mechanisms of insulin resistance in rat models of hypertension and their relationships with salt sensitivity. J Hypertens. 1999 Sep;17(9):1229-37.
53. Leong PKK, Yang L, Landon CS, McDonough AA, Yip KP. Phenol injury-induced hypertension stimulates proximal tubule Na+/H+ exchanger activity. Am J Physiol Renal Physiol. 2006;290:F1543-50.
54. Ferrari P, Ferrandi M, Valentín G. Rostafuroxin: an ouabain antagonist that corrects renal and vascular Na+-K+ ATPase alterations in ouabain and adducin-dependent hipertensión. Am J Physiol Regul Integr Comp Physiol. 2006;290:R529-35.
55. Luft FC. Molecular genetics of salt-sensitivity and hypertension drug metabolism disposition. 2001;29(4):500-04.
56. Barber MO, Barber E, Fox MO, Galvizu K, Sarmiento ME. Efectos de la saralasina y su posterior supresión sobre la presión arterial y la morfofunción renal de ratas normotensas. Rev Cubana Invest Biomed. 1991;10( No. Especial):61.
57. Chaple M, Barber E, Román W, Castillo J, Fox MO, Barber MO. Efectos hemodinámicos de la supresión de propanolol en ratas. Rev Cubana Invest Biomed. 1996;15(2):89-93.
58. Chaple M, Barber E, Fox MO, Castillo J, Barber MO, Chauvín AR. Participación tubular renal en la génesis de la hipertensión arterial primaria. Rev Cubana Invest Biomed. 2003:22(4):217-25.
59. Barber MO, Barber E†, Fox MO. Hipertensión arterial experimental por medio del uso de un bloqueador competitivo de angiotensina II. Rev Cubana Invest Biomed. 2006 Enero;25(1).
60. Comportamiento del volumen sanguíneo corporal en ratas con hipertrofia tubular proximal. Hipertens Riesg Vasc. 2009;26(2):88-9.
61. Hipertensión arterial experimental generada a través del uso de captopril. Nefrología Latinoamericana. 2009;13(1):104.
Recibido: 2 de abril de 2010.
Aprobado: 10 de junio de 2010.
Dra. María Ofelia Barber Fox. Facultad de Ciencias Médicas "Enrique Cabrera". Vento No. 9504 e/ 6 y 10, Altahabana, La Habana, Cuba. Telef: 644 1210 Correo electrónico: mobf@infomed.sld.cu


{kind=link}
{kind=link}