










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Química
versión On-line ISSN 2224-5421
Rev Cub Quim vol.30 no.2 Santiago de Cuba may.-ago. 2018
ARTICULOS
Análisis de metales traza por redisolución anódica con monitoreo potenciometrico redox consistema de mínima instrumentación
Minimal instrumentation anodic stripping potentiometric analysis (spa) of trace metal
Dr. C. Alejandro Baeza-Reyes, Dr. C. Arturo García-Mendoza, MSc. Adrián de Santiago-Zárate
Departamento de Química Analítica, Facultad de Química, UNAM, México, baeza@unam.mx, arturogm@unam.mx, desantiago@hotmail.fr
RESUMEN
Se construyó un sistema de mínima instrumentación y bajo costo para realizar análisis por redisolución anódica , con monitoreo potenciométrico, de una reacción química con Hg(II) en disolución. Se analizaron cationes como Cd(II), Pb(II), Cu(II), Zn(II) y Bi(III) en concentraciones del orden de 10 -5 mol/L y se construyeron las curvas de calibración respectivas, tiempo de transición vs. concentración del catión metálico para utilizarse en el eventual análisis de trazas. Se estudió la influencia del tiempo de depósito durante la polarización del electrodo de trabajo y de la concentración de Hg(II) sobre el tiempo de transición cronopotenciométrico durante el paso de redisolución.
Palabras clave: instrumentación mínima, redisolución, análisis de trazas, microescala.
ABSTRACT
A minimal instrumentation and low-cost equipment for anodic potentiometric stripping analysis was built. The cations Cd(II), Pb(II), Cu(II), Zn(II) and Bi(III) were identified in concentrations around 10 -5 mol/L by calibration plots, such relationships were used for the trace analysis. The effects of deposition time of metals, and their reaction with a fixed concentration of Hg(II) in dissolution over the chronopotentiometric transition time were investigated.
Keywords: minimal instrumentation, stripping, trace analysis, microscale.
INTRODUCCIÓN
La determinación de cationes metálicos a bajos valores de concentración es de gran importancia en química marina, clínica, alimento y ambiental. Los métodos espectrofotométricos por absorción atómica (AA) o análisis por plasma inducido acoplado (ICP son muy sensibles a estos órdenes de concentración, pero requieren de instrumentación de alto costo y mantenimiento. Además, debido a la naturaleza de estas técnicas de análisis, no es posible realizar estudios de especiación química de los metales presentes [1]. Dentro de la amplia gama de métodos electroanalíticos la polarografía diferencial de pulsos permite detectar y especiar metales en un orden de concentración micromolar [2], mientras que el análisis por redisolución anódica con detección por voltamperometría lineal de pulsos ( anodic stripping voltametry, ASV), permite efectuar la determinación de metales micro y nanomolar que corresponde al nivel de trazas o ultratrazas. Este análisis se lleva a cabo en dos etapas. En la primera se ejecuta la pre – concentración por electrodepósito sobre un electrodo estacionario a un valor de potencial de polarización fijo (Epol), de tal manera que se favorezca la electrorreducción del metal en régimen convectivo constante. En la segunda etapa se lleva a cabo la detección por medio de un barrido anódico voltamperométrico lineal [3 – 5].
Los métodos electroanalíticos anteriores requieren de instrumentación de alto costo, así como de medios de reacción y volúmenes de operación de aproximadamente 50,0 mL y de electrodos de trabajo, auxiliares y de referencia de gran tamaño.
Se ha reportado la determinación de metales trazapor análisis de redisolución potenciométrico (ARP)[6]. Un ARP consta de dos etapas [7]:
1. Etapa de pre – concentración o depósito:
Mn+ + ne- + xHg0 -> M0 (Hg)x
2. Etapa de redisolución potenciométrico a corriente nula (potencial de circuito abierto, PCA):
M0 (Hg)x + n/2 Hg2+->Mn+ + (x+n/2)Hg0
En la etapa de pre – concentración se polariza el electrodo a un valor de potencial constante durante un periodo de tiempo de polarización (tpol) en presencia de Hg(II). En dicho periodo el catión analito se reduce y deposita en una amalgama de mercurio sobre el electrodo de trabajo. El tpol es directamente proporcional a la cantidad de analito depositada sobre el electrodo de trabajo. En la etapa de redisolución potenciométrica se determina el potencial de circuito abierto en función del tiempo en el que un agente oxidante en el medio de reacción [en este caso Hg(II)], oxidará al metal amalgamado. La formación del par redox Mn+/M0 (Hg) x proporciona un potencial de celda prácticamente constante. Sin embargo, una vez que se ha oxidado todo el M0 (Hg) x ya no existe más el par redox y ocurre un salto en el valor del potencial de celda, produciendo una curva sigmoidea en un diagrama E vs. t, el cual se puede considerar como un pseudopolarograma [8]. El tiempo entre el término de la polarización del electrodo de trabajo y dicho salto de potencial en la curva se denomina tiempo de transición y es directamente proporcional a la concentración del catión en disolución.
Se ha usado ARP para el análisis de trazas de algunos cationes metálicos como Pb(II), Cd(II), Cu(II) y As(III) en disolución acuosa y en medio amortiguado [9 – 12]. Entre algunas de las ventajas de este método se encuentran la no dependencia de una corriente de electrólisis durante la etapa de redisolución [8], una menor interferencia por sustancias orgánicas absorbibles en el electrodo [13, 14] y un tiempo de análisis relativamente corto y comparable con técnicas como el análisis por redisolución voltamperométrico (ARV). Al igual que el ARV, el ARP descrito utiliza potenciostatos y generadores de señales, celdas y electrodos de alto costo, así como volúmenes considerables de medios de reacción, analitos y muestras.
MATERIALES Y MÉTODOS
La figura 1 muestra el equipo empleado para el electrodepósito y la detección potenciométrica del PCA durante el paso de la redisolución, donde (1) corresponde al micro polarógrafo de mínima instrumentación diseñado para la imposición de la perturbación eléctrica; (2) es el teléfono móvil con equipado con cronometro y con un programa de procesamiento de datos, aunque su empleo es opcional y puede ser remplazado por una computadora (3). En (4) se aprecia una agitador magnético construido también con materiales de bajo costo; en (5) se muestra la jeringa de insulina desechable utilizada para la dosificación de los volúmenes que fueron colocados en (6) el frasco de plástico trihoradado (con capacidad máxima de 5 mL) para celda de análisis. Los electrodos utilizados fueron (7) ER: de Ag0 |AgCl,KCl sat.|| ; (8) ET, disco de carbón vítreo, d = 0,3 mm y (9) EA construido con una barra de grafito número 2, HB. Finalmente, todas las disoluciones fueron homogenizada con ayuda de una barra de agitación de teflón.
La figura 2 muestra el arreglo de celda con tres electrodos que fue conectado a dicho equipo para el análisis por ARP . Se usó un ER de Ag0 |AgCl,KCl sat.||, un ET de disco de carbón vítreo (d = 0,3 mm) y un EA barra de grafito número 2, HB. Al inicio de cada determinación los ET fueron recubiertos, con una capa de mercurio por electrodepósito a partir de una disolución de Hg(II) 25 mg/L, como lo describe Jagner, D. [8]. Se utilizó un medio amortiguado ácido conductor de NaCl 0,5 mol/L y HCl 0,05 mol/L.
Para la determinación de las curvas de calibración se utilizaron 5,0 mL del medio de reacción en la celda, 0,1 mL de Hg(II) 2,5 mmol/L y alícuotas de 0,1 mL de disolución estándar 0,5 mmol/L del catión a analizar en cuestión. Se impuso un Epol = -1,15V vs. ER durante cuatro minutos con agitación constante. Posteriormente se registró el cronopotenciograma a corriente nula, EPCA = f(t). Se ensayaron tiempos de electrodepósito de 8 y 16 min.


RESULTADOS Y DISCUSIÓN
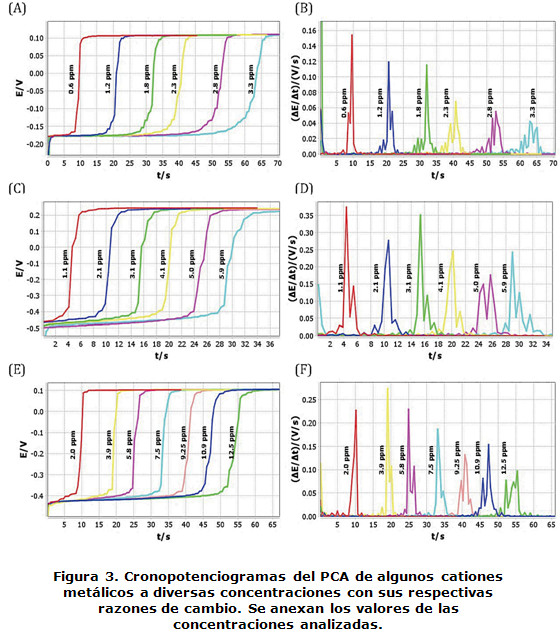
En la figura 3 se muestran los registros típicos de cronopotenciogramas del PCA para (A) Cu(II), (C) Cd(II) y (E) Pb(II), así como sus respectivos cocientes del valor del PCA con respecto al tiempo, utilizado para determinar el tiempo de transición, t, en intervalos de 2 a 9 ppm para (B) Cu(II), (D) Cd(II) y (F) Pb(II). Se ha informado que t es directamente proporcional a la concentración en disolución, de acuerdo a los trabajos de Aoki y Osteryoung [18]. Los valores de las concentraciones analizadas en cada caso se anexan al lado de los registros.
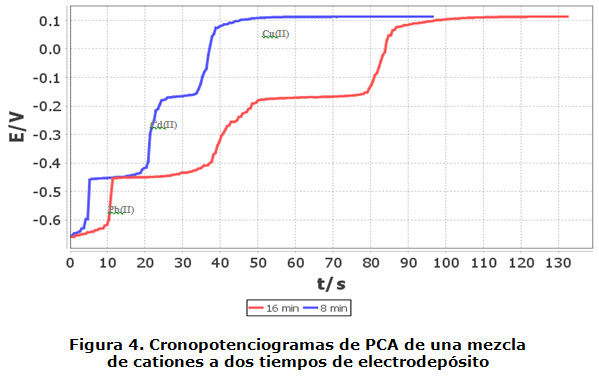
En la figura 4 se muestra el análisis por ARP de una mezcla de cationes, en donde se observa que existen tantos tiempos de transición como cationes presentes hay en la disolución. Tales tiempos de transición se encuentran lo suficientemente separados unos de otros como para ser identificados inequívocamente. Los datos obtenidos indican una proporcionalidad directa entre el tiempo de transición y el tiempo de electrodepósito. Para un electrodepósito de ocho minutos se tienen tiempos de 5,5 s, 21,5 s y 37 s; mientras que para 16 min se obtienen tiempos de 10,5 s, 39 s y 84 s, tales valores prácticamente se duplican al hacerlo con el tiempo de electrodepósito.
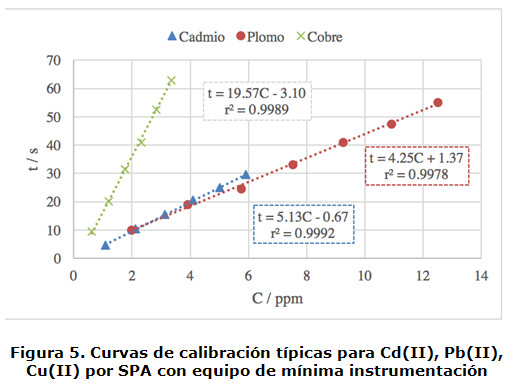
Las curvas de calibración generadas se muestran en la figura 5 a modo de ejemplo para Pb(II), Cd(II) y Cu(II). Los valores de r2 de la regresión lineal son siempre mayores que 0,99 en todos los casos. Los resultados indican que, bajo las condiciones de análisis, el método es más sensible a cobre seguido por plomo y cadmio, para los cuales se obtuvo una sensibilidad semejante.
Adicionalmente se realizaron repeticiones para obtener los parámetros estadísticos de correlación y de los cationes analizados [19]. Se observó que los valores de la incertidumbre relativa a la pendiente de las rectas de calibrado, nunca fueron superiores al 5 %, lo que indica que existe un mínimo grado de dispersión de los puntos de datos experimentales alrededor de la línea de regresión lineal. Los límites de detección encontrados fueron de 0,17, 0,10, 0,44, 0,67 y 0,13 ppm para Cu(II), Cd(II), Pb(II), Bi(III) y Zn(II) respectivamente. En todos los casos analizados, el coeficiente de correlación lineal, r, fue mayor a 0,999 2.
CONCLUSIONES
Se determinó exitosamente la existencia de cationes metálicos, tales como Cu(II), Cd(II), Pb(II), Bi(III) y Zn(II), en concentraciones del orden de 10 ppm o incluso menores, mediante la técnica de ARP en condiciones de microescalamiento empleando materiales de bajo costo, lo que implica una menor generación de residuos y un menor coste operativo. Bajo las condiciones de trabajo se observó una dependencia lineal analíticamente aceptable, entre el tiempo de transición de cada catión metálico durante el proceso de redisolución y la concentración de dicha especie química.
REFERENCIAS BIBLIOGRÁFICAS
1. HARRIS D. C. "Quantitative Chemical Analysis" 5ta edición. Nueva York: W. H. Freeman and Company, 1999. ISBN 9781464135385.
2. FLORES VÉLEZ, L. M.; GUTIÉRREZ RUIZ, M. E.; REYES SALAS, O.; HEYDRYCH, C.; BAEZA REYES, A. "Speciation of Cr(VI) and Cr(III) in soil extract with polarographic methods". International J. Environ. Anal. Chem. 1995, 61, 177–187. ISSN 0306-7319.
3. VÁZQUEZ, G. F.; ELIAS, D. M.; AGUAYO, J. E. C.; BAEZA REYES, A.; SHARMA, V. K. "Trace metal species in aquatic samples of Tabasco Lagoons, Mexico". Environmental International. 1996, 22, 1–6. ISSN 0160-4120
4. SÁNCHEZ GONZÁLEZ, E.; BAEZA REYES, A.; GARCÍA MENDOZA, A. "Modelo básico de una metodología para la determinación y extracción de mercurio en muestras simuladas de fitorremediación con Phaseoulus vulgaris". Rev. Cub. Quim. 2011, 23, 54–57. ISSN 2224-5421.
5. MARRIGO NEGRETE, J.; PINEDO HERNÁNDEZ, J.; BAEZA REYES, A. "Optimization of the electrodeposition conditions for mercury removal from vegetal biomass with response Surface methodology". Port. Electrochim. Acta. 2013, 31 (2), 107 – 117. ISSN 1647-1571
6. WORSFOLD, P.; TOWNSHEND, A.; POOLE, C. F. Encyclopedia of analytical science. 2 da Edición. Amsterdam: Elsevier, 2005. ISBN 978-0-12-369397-6
7. HUSSAM, A.; COETZEE, J. F. "Potentiometric Stripping Analysis: Theory, Experimental Verification and Generation of Heavy Metals in Groundwaters". Anal. Chem. 1985, 57(3), 581–585. ISSN 1520-6882
8. JAGNER, D. "Instrumental approach to potentiometric stripping analysis of some heavy metals". Anal. Chem. 1978, 50(13), 1924–1929. ISSN 1520-6882.
9. BARANSKI, A. S.; QUON, H. "Potentiometric stripping determination of heavy metals with carbon fiber and gold microelectrodes" Anal. Chem. 1986, 58(2), 407–412. ISSN 1520-6882.
10. JAGNER, D.; JOSEFSON, M.; WESTERLUND, S. "Determination of arsenic (III) by computerized potentiometric stripping analysis". Anal. Chem. 1981, 53(13), 2144–2146. ISSN 1520-6882.
11. STANKOVIC, S.; CICKARIC, D.; MARKOVIC, ,J. "Determination of Pb and Cd in water by potentiometric stripping analysis (PSA)". Desalination. 2007, 213(1-3), 282–287. ISSN 0011-9164
12. MUNOZ, E.; PALMERO, S. "Analysis and speciation of arsenic by stripping potentiometry: a review". Talanta. 2005, 65(3), 613–620. ISSN 0039-9140
13. GRANELI, A.; JAGNER, A.; JOSEFSON, D. M. "Microcomputer system for potentiometric stripping analysis" Anal. Chem. 1980, 52(13), 2220–2223. ISSN 1520-6882.
14. CHOW, C. W. K.; DAVEY, D. E.; HASKARD, M. R.; MULCAHY, D. E.; YEOW, T. C. W. "Potentiometric Stripping Analyses: Using a Flexible, Low-cost Computerized Instrument". J. Chem. Educ. 1994 , 71 (11), 997. ISSN 0021-9584.
15. BAEZA, A.; GARCÍA MENDOZA, A.; VIERNA, L. "Microscale Analytical Potentiometry: Experimental Teaching with Locally Produced Low-Cost Instrumentation". J. Mod. Edu. Rev. 2013, 3(5), 407– 15. ISSN 2155-7993.
16. MARÍN-MEDINA A., GARCÍA-MENDOZA A., DE-SANTIAGO A., BAEZA A.. "Diseño, construcción caracterización de titulaciones microcoulombimétricas acido-base con monitoreo visual y micropotencimétrico: Aproximación metrológica". Rev. Cub. Quim. 2014. 26(2), 26–36. ISSN: 2224-5421.
17. VILASO, J. E.; ARADA, M. A.; BAEZA, A.; CESPEDES, A. "Construction and Metrological Characterization of a Minimal Instrumentation Micropolarograph". Port. Electrochim. Acta, 2016, 34, 309–320. ISSN: 1647-1571
18. AOKI, K.; OSTERYOUNG, J. "Formulation of the diffusion-controlled current at very small stationary disk electrodes". Journal of Electroanalytical Chemistry and Interfacial Electrochemistry. 1984, 160(1), 335–339. ISSN 0022-0728.
19. MILLER, J. N.; MILLER, J. C. Statistics and Chemometrics for Analytical Chemistry. 6 ta Edición. Inglaterra: Pearson Education Limited, Harlow, 2011. ISBN 978-0-273-73042-2
Recibido: 5/09/2017
Aceptado: 15/01/2018
Dr. C. Alejandro Baeza-Reyes, Facultad de Química, UNAM, México, baeza@unam.mx

