










Mi SciELO
 Servicios personalizados
Servicios personalizadosServicios Personalizados
Articulo
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO
Links relacionados
-
 Similares en
SciELO
Similares en
SciELO
Compartir

 Permalink
PermalinkRevista Cubana de Cirugía
versión impresa ISSN 0034-7493
Rev Cubana Cir vol.51 no.1 Ciudad de la Habana ene.-mar. 2012
PRESENTACIÓN DE CASO
Linfoma de Burkitt
Burkitt lymphoma
Dr. Fernando Sierra Arego, MSc. Dr. Carlos Michel López Rodríguez
Hospital Docente Clinicoquirúrgico "10 de Octubre". La Habana, Cuba.
RESUMEN
El linfoma de Burkitt es un tipo de linfoma no Hodgkin, no frecuente en Cuba, pues es endémico de África Central. Diagnosticar uno en nuestro país siempre es significativo, por lo que conocerlo es importante.
Palabras clave: linfoma no Hodgkin, linfoma de Burkitt, oclusión intestinal.
ABSTRACT
The Burkitt lymphoma is a type of non-Hodgkin lymphoma infrequent in Cuba since it is endemic of Central Africa. The diagnosis of one in our country always is significant, thus it is important to know it.
Key words: non-Hodgkin lymphoma, Burkitt lymphoma, intestinal occlusion.
INTRODUCCIÓN
Los linfomas no Hodgkin (LNH) son una proliferación monoclonal neoplásica de células linfoides en localizaciones del sistema inmunitario, que incluyen ganglios linfáticos, médula ósea, bazo, hígado y tracto gastrointestinal.1 La clasificación anatomopatológica de los LNH continúa evolucionando, lo que refleja nuevas consideraciones sobre las células de origen y las bases biológicas de este grupo heterogéneo de enfermedades. El curso clínico de los LNH varía desde formas rápidamente mortales, hasta otras quiescentes e inicialmente bien toleradas. Un cuadro similar a la leucemia puede desarrollarse hasta en el 50 % de los niños y alrededor del 20 % de los adultos con algunos tipos de LNH.2-5
Los LNH son más frecuentes que la enfermedad de Hodgkin. En Estados Unidos se diagnostican anualmente alrededor de 50 000 nuevos casos en todos los grupos etarios, y su incidencia aumenta con esta. Se desconoce su causa, aunque, al igual que en las leucemias, existen estudios experimentales que sugieren la participación de ciertos virus en algunos linfomas. Por ejemplo, se ha aislado el retrovirus denominado virus del linfoma leucemia de células T humanas (HTLV-I), que parece ser endémico en el sur del Japón, el Caribe, Sudamérica y el sudeste de Estados Unidos.
La incidencia de LNH, sobre todo de los tipos inmunoblástico y de células pequeñas no hendidas (linfoma de Burkitt), está aumentada en los pacientes infectados por el VIH. En estos casos se ha comunicado afectación primaria del SNC y enfermedad diseminada. En alrededor del 30 % de los casos, los linfomas son precedidos de una linfadenopatía generalizada, lo que sugiere que la estimulación policlonal de los linfocitos B precede a la formación del linfoma.4,5
Las reordenaciones del oncogén c-myc son características de algunos linfomas asociados al SIDA. La respuesta a la quimioterapia es posible, pero también son habituales la toxicidad y las infecciones oportunistas, lo que provoca una supervivencia corta.
La Formulación de Trabajo (Working Formulation) clasifica los LNH en las categorías pronósticas con implicaciones terapéuticas siguientes: (nota: las denominaciones pronósticas se basan en los datos de supervivencia de pacientes tratados antes de 1980, y pueden no reflejar con precisión los resultados obtenidos en pacientes que se someten a terapias modernas, como los que se describen en tratamiento, más adelante):
- Linfomas de bajo grado (38 %): difuso linfocítico de células pequeñas, folicular de células pequeñas hendidas, y folicular mixto de células pequeñas y grandes.
- Linfoma de grado intermedio (40 %): folicular de células grandes, difuso de células pequeñas hendidas, difuso mixto de células pequeñas y grandes, y difuso de células grandes.
- Linfomas de alto grado (20 %): linfoma inmunoblástico, linfoma linfoblástico, y linfoma de células pequeñas no hendidas (tipo Burkitt y no Burkitt).
- Otros linfomas (2 %): linfomas compuestos, micosis fungoide, histiocítico verdadero, otros y tipos inclasificables.
Una nueva clasificación anatomopatológica, la clasificación Revised European-American Lymphoma (REAL) se ha introducido recientemente y se está adoptando de manera gradual. Esta clasificación es útil para identificar entidades no reconocidas en la Formulación de Trabajo, y presenta la característica especial de incorporar rasgos inmunofenotípicos, genotípicos y citogenéticos en las categorías diagnósticas.6,7
Entre los linfomas nuevos más importantes se encuentran los tumores linfoides asociados a mucosas (MALT), el linfoma de células del manto, una enfermedad de mal pronóstico que se clasificaba previamente como un linfoma difuso de células pequeñas hendidas, y el linfoma de células grandes anaplásicas (linfoma Ki-1).8
El linfoma de Burkitt es un linfoma de células pequeñas no hendidas. Es raro en Estados Unidos, pero es endémico en África Central. Puede presentarse en la infancia como una masa mandibular u ovárica de crecimiento rápido. Con mayor frecuencia, aparece como una enfermedad abdominal voluminosa, que se origina a menudo en la región de la válvula ileocecal. En los adultos puede ser voluminosa y generalizada, frecuentemente con afectación masiva del hígado, el bazo y la médula ósea. La afectación cerebral y del líquido cefaloraquídeo (LCR) se observa a menudo en el momento del diagnóstico, o en el caso de linfomas recidivantes.2,7
La anatomía patológica demuestra un índice mitótico elevado y un patrón en cielo estrellado de linfocitos malignos rápidamente proliferativos. La enfermedad se asocia de forma estrecha al virus de Epstein-Barr (VEB) en el linfoma endémico; sin embargo, no está claro que este desempeñe un papel etiológico. El linfoma de Burkitt presenta rasgos citogenéticos característicos, generalmente t,8,9 con implicación del oncogén c-myc. El estudio de extensión consiste en la realización de tomografía computarizada (TC) torácica, biopsia de médula ósea, citología del LCR y gammagrafía con galio. El tratamiento debe iniciarse con urgencia, y los estudios de extensión realizarse con celeridad debido al rápido crecimiento del tumor. Con el tratamiento puede aparecer un síndrome de lisis tumoral como consecuencia de la muerte celular rápida.1,10
Un nivel elevado de L-lactato deshidrogenasa (LDH) predice esta complicación, y los pacientes se tratan con hidratación forzada, alopurinol, alcalinización y vigilancia de los electrólitos (para prevenir o tratar la hiperpotasemia, la posible nefropatía por ácido úrico, la disfunción renal aguda, la hipocalcemia y la hiperfosfatemia). La poliquimioterapia en dosis altas y durante poco tiempo presenta tasas elevadas de curación (> 75 %). La profilaxis meníngea resulta esencial. En ocasiones, la enfermedad se reseca por completo antes de la quimioterapia, aunque la terapia enérgica sigue estando indicada.6
La histopatología, el estadio de la enfermedad, y en algunas series, los resultados de los estudios de marcadores de superficie, influyen significativamente en el pronóstico y la respuesta al tratamiento. Los pacientes con linfomas de células T generalmente tienen peor pronóstico que los que padecen LNH de células B, aunque los resultados de las pautas más recientes de tratamiento intensivo reducen estas diferencias. Otros factores que afectan de forma adversa el pronóstico son: mal estado general, edad superior a 60 años, nivel elevado de LDH, masas tumorales voluminosas (diámetro >10 cm) y presencia de más de dos localizaciones extraganglionares de la enfermedad.
Los pacientes con linfomas de alto grado, linfomas linfoblásticos o linfomas de células pequeñas no hendidas (linfoma de Burkitt), incluso aunque estén aparentemente localizados, deben recibir poliquimioterapia intensiva junto con profilaxis meníngea. El tratamiento puede precisar quimioterapia de mantenimiento (linfoblástica), si bien lo más probable es que se alcance la curación.5,10 La curación se alcanza en el 30-50 % de los pacientes con linfomas de grado intermedio o alto que se someten a terapia mieloablativa. En los linfomas de bajo grado, sigue siendo incierto si puede obtenerse la curación mediante el trasplante, aunque la supervivencia es superior a la que presenta la terapia paliativa secundaria aislada.3,8
La tasa de mortalidad del trasplante mieloablativo se ha reducido notablemente al 2-5 % para la mayoría de los procedimientos autólogos, y a menos del 15 % para la mayor parte de los procedimientos alogénicos.
Una nueva área de investigación es el papel del trasplante autólogo como tratamiento de primera elección en el diagnóstico inicial. Los pacientes de alto riesgo pueden identificarse y seleccionarse para recibir dosis de intensificación mediante el empleo del índice pronóstico internacional (IPI). Los datos preliminares sugieren un incremento de la tasa de curación.
Una secuela tardía de la quimioterapia estándar en altas dosis es la aparición de segundas neoplasias, sobre todo de mielodisplasias y leucemias mieloblásticas agudas. La combinación de quimioterapia y radioterapia aumenta este riesgo, si bien su incidencia continúa siendo próxima al 3 %.4,9
PRESENTACIÓN DEL CASO
Motivo de consulta (MC): dolor abdominal y vómitos.
Historia de la enfermedad actual (HEA): paciente del sexo femenino, de 36 años de edad, que hace más o menos 30 días presenta dolor en el epigastrio y vómitos en número de 5 o 6 por día, tratada en diferentes lugares por otras enfermedades. Hace 15 días esta sintomatología se ha intensificando, y hace cerca de 5 días se ha asociado la no expulsión de heces ni gases por el recto, además de distensión abdominal, por lo que fue traída a nuestro centro para estudio y tratamiento.
- Antecedentes patológicos personales (APP): HTA (por lo que tiene indicado captopril 25 mg 1 tab c/8 h, clortalidona 25 mg 1 tab/día).
- Antecedentes patológicos familiares (APF): no refiere.
- Alergia a medicamentos: no refiere.
- Operaciones anteriores: 2 cesáreas.
- Hábitos tóxicos (HT): no refiere.
- Examen físico:
- Mucosas: ligeramente secas e hipocoloreadas.
- Lengua: seca.
- Tejido celular subcutáneo (TSC): obesa.
- Abdomen: distendido, fundamentalmente en hemiabdomen superior (HAS), palpación dolorosa, timpánico a la percusión, ruidos hidroaéreos disminuidos.
- Aparato cardiovascular (ACV): ruidos taquicárdicos, frecuencia cardíaca (FC): 113/min, TA 150/90 mmHg.
- Indicación diagnóstica (ID): oclusión intestinal mecánica alta.
- Complementarios:
- Hemoglobina (Hb): 85 g/L, hematócrito (Hto): 026 %, coagulación: 9 min y sangrado 1 min, grupo y factor (G y F): O+.
- Electrocardiograma (EKG): taquicardia sinusal.
- Rayos x de tórax postero-anterior (PA): sin alteraciones.
- Rayos x de abdomen simple de pie: múltiples niveles hidroaéreos.
- Rayos x de abdomen simple acostado: imágenes sugestivas de edema interasas.
Tratamiento: quirúrgico.
Informe operatorio: incisión media infraumbilical, que después, por necesidad, se amplía a la media supraumbilical, se abre por planos hasta la cavidad, se localizan estructuras y se encuentra gran distensión de asas yeyunales. Se continúa revisando, y se encuentra la zona de oclusión a punto de partida de una invaginación de cabeza invaginante proximal y una masa tumoral de consistencia firme, carnosa, lisa y redondeada, de unos 8 cm de diámetro (Fig. 1). No es posible la desinvaginación, y se realiza la resección en cuña de la lesión con anastomosis primaria término terminal (T-T) en un plano. A unos 40 cm de esta zona, y distalmente, se encuentra otra lesión tumoral de unos 6 cm que ocluye la luz parcialmente, con iguales características de la anterior. Se decide resecar y realizar sutura primaria T-T en un plano. Durante el acto quirúrgico se encuentra peristalsis aumentada y mantiene ligera distensión, la serosa se aprecia "acartonada" y con cambios en la coloración. Tras revisar la hemostasia y revisar todo el resto de la cavidad abdominal, se cierra en un plano con puntos totales y piel.
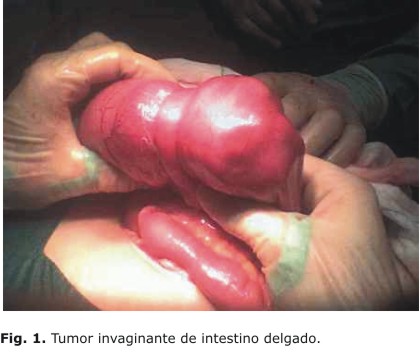
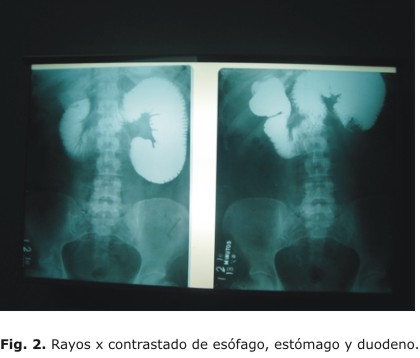
Evolución posquirúrgica: paciente que evoluciona satisfactoriamente, y después de 10 días de ingreso, se decide alta médica. Una semana después la paciente acude con un cuadro oclusivo similar al anterior, se decide realizar un estudio contrastado (rayos x de esófago, estómago y duodeno), y se encuentra una obstrucción al paso del contraste en proyección de asa intestinal yeyunal (Fig. 2), por lo que se confirma el diagnóstico oclusivo y se decide reintervenir.
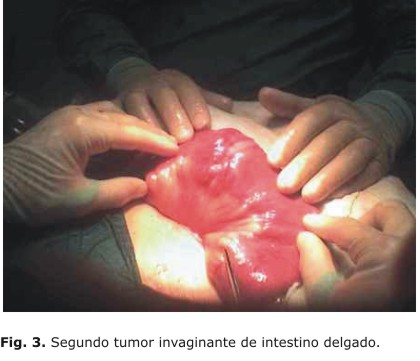
Informe operatorio: se realiza abertura de la cavidad por zona de herida anterior, la cual se amplía un poco más, se encuentran múltiples adherencias muy fibrosas desde asas delgadas iliales a la pared y sutura peritoneal, las cuales se liberan, se continúa la exploración, y se encuentra, a 40 cm del asa fija yeyunal, una masa tumoral invaginante de iguales características que las anteriores, pero más pequeña, de unos 5 cm creciendo hacia la luz, que condiciona la oclusión (Fig. 3). Se realiza resección con anastomosis primaria T-T en un plano, se revisa nuevamente toda la cavidad abdominal, y se mantienen los cambios de la serosa encontrados en la operación anterior, sin otras alteraciones. Se decide el cierre de la cavidad con puntos totales y cierre de piel.
Evolución posoperatoria: paciente que evoluciona satisfactoriamente, y después de 10 días en el hospital, se decide dar el alta.
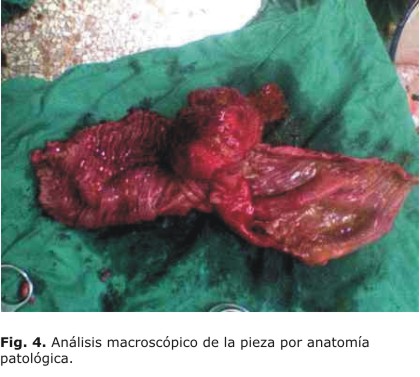
Tras la espera por el resultado del análisis anatomopatológico de la muestra (Fig. 4), la cual se tuvo que realizar —una parte de ella— en el Instituto Nacional de Oncología y Radiobiología (INOR) de la capital, para estudio histoinmunoquímico, se recibe lo siguiente:
- Pieza T 50 500 por técnicas inmunohistoquímica (IHQ) en cortes de parafina: linfoma de Burkitt.
- CD20(+), CD10(+), CD3(-), TDT(-), KI-67 (95 %) (firmado por el doctor Jiménez, del INOR). Otros complementarios de interés: VIH negativo.
- Eritrosedimentación: 100 mm/h.
Evolución actual: paciente que tras una evolución satisfactoria de sus intervenciones, está recibiendo tratamiento complementario con poliquimioterapia en INOR.
DISCUSIÓN
Esta paciente durante los años anteriores al diagnóstico de la enfermedad, excepto por la HTA, no presentó ningún otro problema que hiciera pensar en esta enfermedad de inicio, sin APF, ni otro indicio de importancia que la relacionara. En el momento del diagnóstico de la oclusión intestinal, se pensó, de inicio, en la posibilidad de que la causa fuera por bridas, como una de las causas más frecuentes y teniendo en cuenta las operaciones anteriores. Lo otro a destacar es la evolución sincrónica de los tumores,3 localizados en las 2 intervenciones en el intestino delgado, y específicamente en yeyuno, y que solo medió entre una y otra operación 18 días, con un marcado carácter agresivo en cuanto a evolución de la enfermedad, así como también, la aparición de adenopatías en número de 2 en región axilar derecha y cadena ganglionar cervical derecha, posterior a todo el tratamiento quirúrgico realizado y toda su evolución hasta su alta médica. Además, es importante mencionar que hasta el momento, con los complementarios de sangre que se le han realizado, no se han encontrado causas de inmunodepresión que pudieran relacionarse con la aparición o desarrollo de la enfermedad.
Por todo ello, el caso que nos ocupa realmente tiene, además de importancia por lo infrecuente en nuestro país, la sugerencia de su reporte a la comunidad médica como algo que debe ser conocido por los que de una manera u otra pueden atender a estos pacientes (clínicos, oncólogos, patólogos y cirujanos).
REFERENCIAS BIBLIOGRÁFICAS
1. Beers HM, Porter SR, Jones VT, Kaplan LJ, Berkwits M. Manual Merck. 11na. Edición. España: Elsevier; 2007. p. 1224-6.
2. Harrison TR. Principios de Medicina Interna. 16ma. edición. En: Parte V. Oncología y hematología. Manifestaciones clínicas, tratamiento y pronóstico de las distintas neoplasias linfoides malignas (en español). Harrison online en español. México DF: McGraw-Hill; 2006 [citado el 13 de junio de 2008]. Disponible en: http://harrisonmedicina.com/content.aspx?aID=65919&searchStr=linfoma+de+burkittt#65919
3. Liu D, Shimonov J, Primanneni S, Lai Y, Ahmed T, Seiter K. T 8, 14, 18: a 3-way chromosome translocation in two patients with Burkitt's lymphoma/leukemia. Mol Cancer. 2007;6:35.
4. Cecil RLF, Russell FS, LLoyd H, Wyngaarden JB. Tratado de medicina interna. 23rd ed. Philadelphia: Saunders Elsevier; 2007. p. 1411.
5. Horning SJ. Hodgkin's lymphoma. In: Abeloff MD, Armitage JO, Niederhuber JE, Kastan MB, McKena WG, eds. Clinical Oncology. 4th ed. Philadelphia: Elsevier Churchill Livingstone; 2008. p. 2274-80.
6. Bacon CM, Du MQ, Dogan A. Mucosa-associated lymphoidtissue (MALT) lymphoma: a practical guide for pathologists. J Clin Pathol. 2007;60:361-72.
7. Marcus R, Imrie K, Belch A, Cunningham D, Flores E, Catalano J, et al. CVP chemotherapy plus rituximab compared with CVP as first-line treatment for advanced follicular lymphoma. Blood. 2005;105:1417-23.
8. Olivieri A, Santini G, Patti C, Chisesi T, De Souza C, Rubagotti A, et al. Upfront high-dose sequential therapy (HDS) versus VACOP-B with or without HDS in aggressive non-Hodgkin's lymphoma: long-term results by the NHLCSG. Annals of Oncology. 2005;16:1941-8.
9. Meremikwu MM, Ehiri JE, Nkanga DG, Udoh EE, Ikpatt OF, Alaje EO. Socio-economic constraints to effective management of Burkitt's lymphoma in south-east Nigeria. Tropical medicine and international Elath; 2005 (en prensa).
10. Hainsworth JD, Litchy S, Shaffer DW, Lackey VL, Grimaldi M, Greco AF. Maximising therapeutic effect of rituximab: maintenance treatment vs. re-treatment at progression in patients with indolent non-Hodgkin's lymphoma- a randomized phase two trial of the Minie Pearl research network. Journal of clinical oncology. 2005;23(6):1088-95.
Recibido: 19 de agosto de 2010.
Aprobado: 8 de noviembre de 2010.
Fernando Sierra Arego. Hospital Docente Clinicoquirúrgico "10 de Octubre". Calzada de 10 de Octubre no. 130, entre Alejandro Ramírez y Agua Dulce, municipio Cerro. La Habana. Cuba. Correo electrónico: fernando.sierra@infomed.sld.cu

