










 Servicios personalizados
Servicios personalizados Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por email Citado por SciELO
Citado por SciELO  Similares en
SciELO
Similares en
SciELO 
 Permalink
PermalinkIntroducción
En niños y adolescentes los tumores de corteza suprarrenal son infrecuentes. Se caracterizan por una presentación heterogénea y patogenia aún entendida parcialmente. Tienen la particularidad de que pueden determinar un efecto deletéreo sobre el crecimiento del niño debido a una maduración sexual y somática precoces y patológicas.1
Tienen una incidencia de 0,5 a 2 casos por cada millón de habitantes al año.2) La relación entre mujeres y hombres es de 2,5: 3,1. Se presenta en dos picos: en la primera década y entre la cuarta y la quinta décadas de la vida. La incidencia en edades pediátricas es de 0,2-0,3 casos por cada millón de niños al año.3) Aproximadamente, el 75 % de estos casos se observan en los primeros 5 años y afectan a las niñas con mayor frecuencia que a los niños.4
En EE.UU representa el 0,2 % de todas las neoplasias informadas anualmente, con una incidencia de 0,2-0,3 casos por 1 millón de habitantes por año.5) En menores de 19 años, según datos de Surveillance, Epidemiology and Endresults,(SEER) perteneciente al National Cancer Institute, se estima que solamente el 1,0 % de los procesos de la infancia son carcinomas, siendo apenas un 0,1 % tumores de corteza suprarrenal.6
No obstante, existen variaciones geográficas como en el sudeste de Brasil, donde la presencia es aproximadamente 15 veces superior al reportar 3,4 casos por millón de niños menores de 14 años por año. En 1990 se fundó el International Pediatric Adrenocortical Tumor Registry (IPACTR) en el St. Jude Children's Research Hospital de Memphis, EE.UU y el hospital de clínicas de Curitiba, Brasil con el objetivo de ganar en experiencia al disponer de un mayor número de casos clínicos, muestras de laboratorio, tejidos y estudios moleculares, además de investigar nuevas vías terapéuticas. En 2002 el Children's Oncology Group (COG) creó el Comité de Tumores Raros, donde incluyó un subcomité para el estudio de los tumores de corteza.1
En el caso de Cuba la incidencia de carcinoma corticosuprarrenal es baja, y representa solo el 6 % de los tumores adrenales en la infancia.7) Según el Registro Nacional de Cáncer, la tasa es de 0,4 por cada 100 000 habitantes.8
La causa es desconocida, por lo que recientemente se han utilizado para este fin marcadores moleculares como los factores de crecimiento 1 y 2, semejantes a la insulina (IGF-1) e (IGF-2), y genéticos (estudio de genes 11p, 13q o 17p) involucrados en este tipo de entidad.3) Además, se ha descrito su asociación con síndromes hereditarios tales como el síndrome de Lynch,9,10 el síndrome de Li- Fraumeni,11,12 la adenopolimatosis familiar,10 la neoplasia endocrina múltiple (MEN) tipo 1,13,14 y el síndrome de Beckwith-Wiedermann.15,16) En edades pediátricas es comúnmente asociado con alteraciones genéticas y/o epigenéticas, representado por anormalidades a nivel del cromosoma 11p y mutaciones de la línea germinal TP53 en el 50-90 % de los casos.17
La clínica de los carcinomas adrenales debe ser considerada en tres categorías: los síntomas debido a la propia masa adrenal, los debido a la diseminación local, regional o a distancia, y los de hiperfunción endocrina.18
Un número creciente de casos, aproximadamente entre el 10-15 % se diagnostica dentro del grupo de masas suprarrenales descubiertas incidentalmente (incidentalomas). Sin embargo, la probabilidad de que un incidentaloma suprarrenal sea un carcinoma adrenocortical es baja. Aproximadamente del 50 al 60 % tienen un exceso clínico de hormonas. En la mayoría se observa hipercortisolismo (síndrome de Cushing) o síndromes mixtos de Cushing y virilización. El exceso de andrógenos puros es menos frecuente, mientras que el exceso de estrógenos o mineralocorticoides es muy raro.15
Los síntomas inespecíficos de una masa abdominal incluyen malestar abdominal (náuseas, vómitos, plenitud abdominal) o dolor de espalda.17 Los síntomas clásicos asociados a la malignidad, como pérdida de peso, sudores nocturnos, fatiga o fiebre, rara vez están presentes.15
Se presenta un caso clínico con el objetivo de describir las características clínicas, los procederes diagnósticos y terapéuticos de un paciente con carcinoma adrenal en edad pediátrica.
Presentación del caso
Escolar masculino de 8 años de edad nacido a término producto de un parto distócico por cesárea, sin antecedentes prenatales de interés. Es remitido por su médico de la familia a la Consulta de Endocrinología del Hospital Pediátrico Provincial “Octavio de la Concepción y la Pedraja”. La madre refirió que el niño hacía dos años presentaba vello sexual púbico y aumento del pene en longitud y grosor, aunque no había sido atendido por este motivo con anterioridad. Se decide su ingreso para el estudio por pubertad precoz y se le indican los exámenes complementarios correspondientes.
Como antecedentes patológicos personales se destaca el asma bronquial, aunque no requiere tratamiento intercrisis. Como antecedentes familiares se reporta que el padre padece cáncer (linfoma cervical), actualmente con seguimiento oncológico. Su tía materna está operada de cáncer de mama y su tío materno fue operado de cáncer de colon. Su abuela materna presenta hipertensión arterial y la tía materna, diabetes mellitus tipo 2. Niega antecedentes de administración de gonadotropina coriónica humana (HCG) o andrógenos.
A su ingreso se registró un peso de 27,5 kg y talla de 130 cm, lo que se corresponde con una valoración nutricional según las tablas de P/E y T/E 75-90p y de P/T 50-75p, IMC 16,27 kg/m2 (50-75p). Presenta una discreta aceleración en la velocidad de crecimiento pasando del percentil 25 al 50-75p con adecuado incremento ponderal y signos vitales normales. En el examen físico se detectó voz gruesa, sin cambios en la composición corporal, presencia de vello axilar y vello sexual púbico estadio III de Tanner. Mostró ausencia de vello en otras áreas corporales andrógenodependientes, genitales externos estadio III de Tanner, pene de 7,5 cm de longitud y 2 cm de diámetro, piel de las bolsas escrotales enrojecida, testes descendidos con un volumen testicular derecho 3 ml y el izquierdo de 4 ml medidos con el orquidómetro de Prader. El resto del examen físico y endocrino dio negativo.
Exámenes complementarios
Generales
- Hemograma completo: Hb 136 g/L, diferencial normal.
- Eritrosedimentación: 11 mm/h.
- Coagulograma: 80 % de macroplaquetas. El resto es normal.
- Glucemia: 5,0 mmol/L.
- Colesterol: 4,0 mmol/L.
- Triglicéridos: 1,0 mmol/L.
- TGP: 5 U/L.
- TGO: 10 U/L.
- Creatinina: 60 umol/L.
- Urea: 4,9 umol/L.
- Ácido úrico: 192 umol/L.
- Bilirrubina directa: 1,9 umol/L.
- Bilirrubina indirecta: 9 umol/L.
- Ionograma: Na: 139mmol/L, K: 4,4 mmol/L, Cl: 98,2 mmol/L, Cai: 1,11 mmol/L.
Hormonales
- FSH: 0,01mUI/mL, VR: 1,5-12,4 mUI/mL.
- LH: 0,03 mUI/mL, VR: 1,7-8,6 mUI/mL.
-T estosterona total: 36 ng/mL (elevada), VR: 2,19-8,30 ng/mL.
- 17OH: 55ng/ml (elevada), VR: 0,00-40,15 ng/mL.
- DHEA-S: 1200 Ug/mL(elevada), VR: 0,003- 270 Ug/mL.
- Cortisol: 327 nmol/L, VR: 171-536 nmol/L.
- El resto de los estudios hormonales dieron negativos.
Imagenológicos
- Edad ósea꞉ acelerada, correspondiente con 11-12 años.
- Rx selectivo de silla turca: normal.
- Ultrasonografía (US) abdominal꞉ hígado, vesícula, páncreas y riñones normales. Imagen hipoecogénica a nivel de la glándula suprarrenal izquierda con medidas de 52x50 mm de contornos regulares y bien definidos, de estructura interna heterogénea, con múltiples calcificaciones.
- US de testículos: testículo derecho 20x8 mm, testículo izquierdo 23x8 mm, cabeza del epidídimo 6 mm el derecho y 7 mm el izquierdo, glándula de tamaño y ecogenicidad normales, sin lesión focal.
- Tomografía computarizada (TC) contrastada de abdomen꞉ imagen tumoral hipodensa (17-25UH) de 55 x 49 mm, de textura interna heterogénea, con múltiples calcificaciones salteadas en su interior. Lesión de contornos regulares y bien definidos de localización suprarrenal izquierda, que producen compresión del polo renal superior, con desplazamiento posterior del mismo, de la cola pancreática y la vena esplénica hacia delante. Ligero realce tumoral pos contraste, discreta hidronefrosis izquierda pos compresiva. No se visualizan adenopatías ni lesiones en el resto de las estructuras visibles (Fig. 1).
- No se indican estudios genéticos por no disponibilidad.
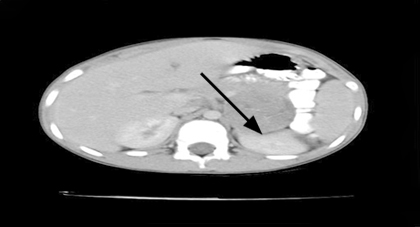
Evolución intrahospitalaria
Se emite como diagnóstico un tumor suprarrenal virilizante izquierdo y se decide su traslado para el Servicio de Cirugía para tratamiento quirúrgico.
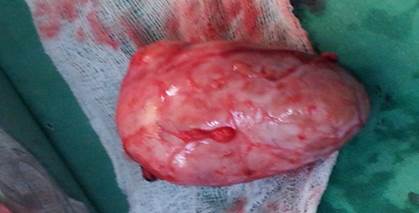
El paciente se sometió a cirugía y se le realizó una laparotomía a través de incisión transversa supraumbilical izquierda. Se localizó un tumor suprarrenal izquierdo en el retroperitoneo, se disecó y se ligaron los vasos sanguíneos. Se realizó exéresis completa de este y una linfadenectomía regional, la cual se envió a Anatomía Patológica junto a un segmento de grasa perirrenal anterior. Se revisó el riñón contralateral que no mostró alteraciones. Sin embargo, el riñón izquierdo mostró discreta hidronefrosis.(Fig. 2)
Se medicó profilácticamente con hidrocortisona durante el pre, el trans y el posoperatorio y no se reportaron complicaciones. La evolución posoperatoria fue adecuada.
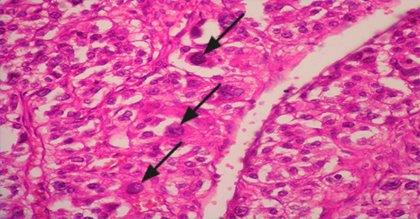
El estudio histopatológico, según escala de Weiss,15 reportó un carcinoma de la glándula suprarrenal izquierda, productor de andrógenos de 6x5x4 cm. Estaba constituido por células fasciculares y reticulares fundamentalmente con una cápsula en todo su espesor y la grasa periglandular. No se encontró infiltración de la cápsula perirrenal. Se encontraron zonas de calcificación distróficas tumorales, necrosis y hemorragias (Fig. 3).
El paciente fue trasladado para el Servicio de Oncología. Se le indicaron los complementarios generales e imagenológicos con resultados normales. Se descartaron lesiones metastásicas y hormonales evolutivas normales.
Exámenes complementarios
- FSH: 1,2 mUI/mL.
- LH 1,3 mUI/mL.
- Testosterona: 2,75 ng/mL.
- 17OH: 3,91ng/mL.
Se realizó una estadificación, según el sistema de clasificación TNM del Comité Americano Conjunto de Cáncer.19) Se emitió como diagnóstico definitivo un carcinoma corticosuprarrenal virilizante izquierdo estadio 2 (T2 N0 M0). En relación al protocolo de tratamiento, no correspondía por su estadio clínico quimioterapia por tratarse de un tumor con alto grado de malignidad. Se decidió por un tratamiento con cisplastino, etopóxido y doxorrubicina como tratamiento paliativo como quimioterapia previa a recidiva tumoral. No hubo tratamiento con mitotane por falta de disponibilidad.
Discusión
El carcinoma de la corteza suprarrenal representa desafortunadamente una de las neoplasias endocrinológicas más agresivas.20 De ellos, el 79 % son funcionantes.21) En niños es infrecuente y solo representa el 6 % de los tumores adrenales en la infancia. La supervivencia es el doble que en adultos y la mayoría son hormonalmente activos, (alrededor del 90 %).7,22
Con relativa frecuencia cuando se realiza el diagnóstico del carcinoma corticosuprarrenal ya se encuentra en estadios avanzados. Esto se debe en gran medida a la tendencia que tiene a invadir las estructuras vasculares, lo que origina una metastización precoz.18) En el niño prepúber, la manifestación más típica es el desarrollo de pubertad precoz.21
Se plantea una pubertad precoz periférica (PPP) en este reporte, por lo que es importante descartar aquellas entidades que la producen en el varón. Pueden ser atribuible a las siguientes causas:
Secreción endógena ectópica o autónoma de HCG o LH (hormona luteinizante) por tumores como hepatoblastoma, teratoma, gonadoblastoma, coriocarcinoma o administración iatrogénica de HCG, los cuales pueden aumentar la producción de testosterona en las células de Leydig.
Alteraciones genéticas del receptor (testotoxicosis, síndrome de Mc Cune Albright).
Secreción endógena autónoma de andrógenos de los testículos (tumor de células de Leydig) o glándulas suprarrenales (adenoma, carcinoma, hiperplasia suprarrenal congénita) o por administración exógena iatrogénica de andrógenos o disruptores endocrinos.23
El hipotiroidismo primario y la resistencia primaria al cortisol también pueden ser causa de PPP.24,25
En los varones con PPP es característico el desarrollo progresivo de signos de virilización como el incremento del tamaño del pene, sin un aumento significativo del tamaño testicular. En algunos casos debido a la testotoxicosis, los restos adrenales testiculares, los tumores productores de HCG, entre otros, el volumen testicular puede incrementarse ligeramente (4-8 mL). En cualquier caso es un volumen inadecuado para el grado de desarrollo de los caracteres sexuales secundarios. El desarrollo de signos feminizantes (ginecomastia marcada) es excepcional, pero puede producirse en el contexto de exposición a una fuente externa de estrógenos o en raros casos de tumores testiculares (tumor de células de Sertoli asociado al síndrome de Peutz-Jegher) o adrenales productores de estrógenos.26
Las entidades nosológicas más características responsables de PPP en el varón son la hiperplasia adrenal congénita, el síndrome de McCune-Albright y la testotoxicosis. Sin embargo, los tumores suprarrenales son muy raros. Es por ello que para poder determinar el origen de la producción androgénica se debe precisar los niveles de andrógenos como androstenediona, dehidroepiandrosterona sulfato (DHEAS) y testosterona, los cuales se encontrarán elevados.27
En los casos de carcinomas corticosuprarrenales, la pubertad precoz secundaria a la excesiva producción de andrógenos se evidencia por el incremento del tamaño del pene en los niños. En las niñas por hirsutismo y clitoromegalia, además de pubarquia se manifiesta el sudor apocrino, la aceleración en el crecimiento y la voz ronca.22 Similares hallazgos fueron encontrados en el paciente en cuestión, correspondiéndose con lo reportado en la literatura. Sin embargo, en el varón adulto la hipersecreción androgénica puede pasar desapercibida y manifestarse tan solo por un aumento de peso secundario al efecto anabolizante.28
La mayoría de los carcinomas corticosuprarrenales son esporádicos, pero pueden ser un componente de un síndrome hereditario.29 Los antecedentes de cáncer en familiares de primer y segundo grado son importantes, debiéndose descartar el síndrome de Li Fraumeni que se caracteriza por un patrón de herencia autosómico dominante y expresividad variable, relacionada con alteraciones en el gen TP53.30
Dicha entidad predispone al desarrollo de una amplia variedad de tumores malignos. Los tumores más característicos son los sarcomas de tejido blando (rabdomiosarcoma y otros), osteosarcoma, cáncer de mama en mujeres premenopáusicas, leucemia, linfomas, tumores cerebrales (carcinoma de plexos coroideos, glioblastoma y meduloblastoma) y carcinoma adrenocortical. Dichos tumores pueden presentarse a cualquier edad, incluida la pediátrica. La prevalencia no es bien conocida pues es sin duda una entidad infradiagnosticada. Dado que el seguimiento de familias no ha demostrado aumentar la supervivencia a largo plazo, no existe hasta la fecha un programa de detección de pacientes.22,30
Los carcinomas adrenales presentan a menudo un fallo enzimático en los sistemas de la 11-beta-hidroxilasa, pudiendo presentar también defectos de la 17-alfa-hidroxilasa, 21-hidroxilasa, 3-beta-hidroxiesteroide-deshidrogenasa y delta5-delta4-isomerasa. Por ello, aunque se acepte que la mayor parte son tumores no funcionantes, la realidad es que secretan distintos tipos de precursores que es necesario buscar. No obstante, se ha descrito la secreción de metabolitos de pregnenolona, tetrahidrodeoxicortisol, 11-desoxicortisol, pregnanediol, ocasionalmente testosterona no derivada del metabolismo periférico de la androstenediona, estrona, estradiol y desoxicorticosterona.28 En los estudios hormonales se constataron niveles de 17 hidroxiprogesterona elevados coincidiendo con lo anteriormente expuesto.
Los estudios imagenológicos juegan un rol importante para el diagnóstico de los tumores intrabdominales, destacándose entre ellos la ultrasonografía y la tomografía computarizada, tanto en los pequeños como en grandes masas tumorales.5) El estudio de imagen de elección es la tomografía computarizada (TC), en el que se detectó un tumor adrenal grande (masa suprarrenal mayor de 4 cm, lo cual es predictivo de malignidad), unilateral, heterogéneo, con densidad >20 UH y múltiples calcificaciones. También pueden presentar hemorragias, bordes irregulares, retardo en eliminación de medio de contraste (<50 % en 10 min), invasión a estructuras vecinas, ganglios linfáticos aumentados de tamaño o metástasis a otros órganos.31,32
La resonancia magnética (RM) se utiliza como segunda línea y tiene mayor especificidad sobre todo en lesiones menores de 4 cm al detectar lesiones hipointensas respecto al hígado en T1. La tomografía con emisión de positrones (PET) tiene una alta sensibilidad y especificidad para lesiones malignas, incluso menores de 1,5 cm, además de detectar metástasis.31
Existe una superposición considerable en el ámbito clínico, radiológico y características histológicas del carcinoma y el adenoma adrenocortical que dificulta la toma de decisiones. Debido a la rareza y heterogeneidad del carcinoma adrenal pediátrico, se limitan las evidencias existentes para definir un criterio histopatológico para diferenciar el carcinoma de los adenomas adrenocorticales. La puntuación de Weiss se utiliza ampliamente para evaluar el carcinoma adrenocortical en adultos. La versión modificada y el índice de Wieneke se han propuesto para evaluar el carcinoma adrenocortical en pediatría.3
Los estudios retrospectivos han logrado identificar dos importantes factores pronósticos: la efectividad de la resección quirúrgica y el estadio tumoral. Se han intentado buscar múltiples parámetros morfológicos que permitan predecir el comportamiento de estas lesiones. No obstante, algunos casos con parámetros de mal pronóstico tienen un comportamiento benigno o de muy baja agresividad. Los pacientes que no muestran pruebas de invasión del tejido local o de los ganglios linfáticos, tendrán un mejor pronóstico.33
La cirugía es el pilar del tratamiento, pero incluso después de la completa resección existe un alto riesgo de recurrencia. A pesar de los múltiples enfoques de tratamiento que incluyen el mitotane como droga adrenolítica, la quimioterapia con cisplastino, etoposide y doxorrubicina, y la radioterapia, el pronóstico del carcinoma adrenocortical en edades pediátricas sigue siendo pobre con una supervivencia estimada de 5 años y una tasa de supervivencia que oscila entre el 30-90 %.3) Por esta razón, es que el diagnóstico temprano es la clave para asegurar un mejor pronóstico.15
Conclusiones
Los carcinomas corticosuprarrenales en niños son mayoritariamente funcionantes, constituyendo una de las causas de pubertad precoz periférica. Estos tumores son infrecuentes y agresivos, de ahí la importancia de su diagnóstico precoz para un adecuado tratamiento y mejor pronóstico.
El despistaje de cáncer en familiares de pacientes con síndromes hereditarios asociados a carcinoma corticosuprarrenal, así como la realización de estudios genéticos contribuiría al diagnóstico en estadios iniciales y por ende a una disminución de la mortalidad por esta patología.

